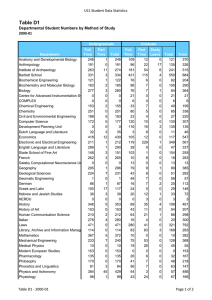
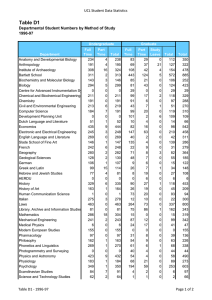
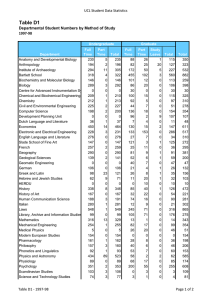
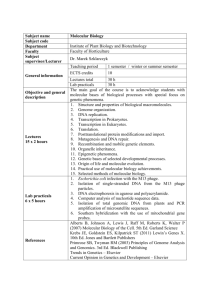
Molecular Biology Published by TSRI Press . Copyright 2005,
advertisement

Molecular Biology Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Yun Yung, Graduate Student, and Jerold Chun, M.D., Ph.D., Professor, Department of Molecular Biology Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. MOLECULAR BIOLOGY 2005 155 DEPAR TMENT OF MOLECULAR BIOLOGY S TA F F Peter E. Wright, Ph.D.* Professor and Chairman Cecil H. and Ida M. Green Investigator in Medical Research Ruben Abagyan, Ph.D. Professor Carlos F. Barbas III, Ph.D.* Professor Janet and W. Keith Kellogg II Chair, Molecular Biology Michael N. Boddy, Ph.D. Assistant Professor Charles L. Brooks III, Ph.D. Professor Monica J. Carson, Ph.D.** Associate Professor University of California Riverside, California David A. Case, Ph.D. Professor Geoffrey Chang, Ph.D.* Associate Professor Jerold Chun, M.D., Ph.D. Professor Lisa Craig, Ph.D.** Assistant Professor Simon Fraser University Burnaby, British Columbia Valerie De Crecy Lagard, Ph.D.** Assistant Professor University of Florida Gainesville, Florida Luis De Lecea, Ph.D. Associate Professor H. Jane Dyson, Ph.D. Professor John H. Elder, Ph.D. Professor Martha J. Fedor, Ph.D.* Associate Professor Richard A. Lerner, M.D., Ph.D.***** President, Scripps Research Lita Annenberg Hazen Professor of Immunochemistry Cecil H. and Ida M. Green Chair in Chemistry James Arthur Fee, Ph.D. Professor of Research Scott Lesley, Ph.D. Assistant Professor of Biochemistry Elizabeth D. Getzoff, Ph.D.**** Professor Tianwei Lin, Ph.D. Assistant Professor David B. Goodin, Ph.D. Associate Professor David S. Goodsell Jr., Ph.D. Associate Professor Joel M. Gottesfeld, Ph.D. Professor Robert Hallewell, D.Phil. Adjunct Associate Professor Jennifer Harris, Ph.D. Assistant Professor of Biochemistry Christian A. Hassig, Ph.D. Adjunct Assistant Professor Mirko Hennig, Ph.D. Assistant Professor John E. Johnson, Ph.D. Professor Clare McGowan, Ph.D. † Associate Professor Duncan E. McRee, Ph.D. Adjunct Associate Professor David P. Millar, Ph.D. Associate Professor Louis Noodleman, Ph.D. Associate Professor Arthur J. Olson, Ph.D. Professor James C. Paulson, Ph.D. †† Professor Vijay Reddy, Ph.D. Assistant Professor Steven I. Reed, Ph.D. † Professor Gerald F. Joyce, M.D., Ph.D.***** Professor Victoria A. Roberts, Ph.D.** Associate Professor University of California San Diego, California Ehud Keinan, Ph.D. Adjunct Professor Paul Russell, Ph.D. Professor Michel Sanner, Ph.D. Associate Professor Harold Scheraga, Ph.D. Adjunct Professor Paul R. Schimmel, Ph.D.***** Ernest and Jean Hahn Professor of Molecular Biology and Chemistry Anette Schneemann, Ph.D. Associate Professor Subhash C. Sinha, Ph.D.* Associate Professor Gary Siuzdak, Ph.D. Adjunct Associate Professor Robyn L. Stanfield, Ph.D. Assistant Professor James Steven, Ph.D. Assistant Professor Raymond C. Stevens, Ph.D.††† Professor Charles D. Stout, Ph.D. Associate Professor Peiqing Sun, Ph.D. Assistant Professor J. Gregor Sutcliffe, Ph.D. Professor John A. Tainer, Ph.D.* Professor Fujie Tanaka, Ph.D. Assistant Professor Elizabeth Anne Thomas, Ph.D. Assistant Professor S E C T I O N C O V E R F O R T H E D E P A R T M E N T O F M O L E C U L A R B I O L O G Y : Toll-like receptors (TLRs) recognize various pathogen-associated molecules and play an important role in innate immune responses. The human TLR3 recognizes double-stranded RNA from viruses and initi- Lluis Ribas De Pouplana, Ph.D. Adjunct Assistant Professor ates an intracellular signaling pathway through the interaction of TIR domains of TLR3 and the adaptor molecule TRIF. The proposed dimer of the TLR3 ectodomain is displayed on the membrane surface with double-stranded RNA from viruses. The crystal structure was determined by Jungwoo Ashok Deniz, Ph.D. Assistant Professor Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Choe, Ph.D., in the laboratory of Ian A. Wilson, D.Phil. 156 MOLECULAR BIOLOGY 2005 James R. Williamson, Ph.D.***** Professor Associate Dean, Kellogg School of Science and Technology Ian A. Wilson, D.Phil.* Professor S TA F F S C I E N T I S T S Aymeric Pierre De Parseval, Ph.D. Karla Ewalt, Ph.D.** Princeton University Princeton, New Jersey Liang Tang, Ph.D.** Burnham Institute La Jolla, California Christopher Baskerville, Ph.D. Ellie Tzima, Ph.D.** University of North Carolina Chapel Hill, North Carolina Konstantinos Beis, Ph.D. Xiang-Lei Yang, Ph.D. Svitlana Berezhna, Ph.D. Dirk M. Zajonc, Ph.D. William Henry Bisson, Ph.D. Lipika Basummalick, Ph.D. Per Bengston, Ph.D. Brian M. Lee, Ph.D. Curt Wittenberg, Ph.D. † Professor Kurt Wüthrich, Ph.D. Cecil H. and Ida M. Green Professor of Structural Biology Maria Martinez-Yamout, Ph.D. Garrett M. Morris, Ph.D. Chiaki Nishimura, Ph.D. Jeffrey Speir, Ph.D. Todd O. Yeates, Ph.D. Adjunct Professor Qinghai Zhang, Ph.D. Assistant Professor Manal Swairjo, Ph.D. Mutsuo Yamaguchi, Ph.D. Xueyong Zhu, Ph.D. Guo Fu Zhong, Ph.D.** Fudan University Shanghai, China SENIOR RESEARCH R E S E A R C H A S S O C I AT E S Sunny Abraham, Ph.D. Fabio Agnelli, Ph.D. Moballigh Ahmad, Ph.D. Alexander Ivanov Alexandrov, Ph.D. Marcius Da Silva Almeida, Ph.D. Pilar Blancafort, Ph.D.** University of North Carolina Chapel Hill, North Carolina David Boehr, Ph.D. David Bostick, Ph.D. Ronald M. Brudler, Ph.D. Lintao Bu, Ph.D. Rosa Maria Cardoso, Ph.D. Justin E. Carlson, Ph.D. A S S O C I AT E S Beatriz Gonzalez Alonso, Ph.D. SERVICE FACILITIES Andrew Barry Carmel, Ph.D. David Barondeau, Ph.D. Ola Blixt, Ph.D. Core Manager, Consortium for Functional Glycomics Kirk Beebe, Ph.D. John Chung, Ph.D. Manager, Nuclear Magnetic Resonance Facilities Brian Collins, Ph.D. Gerard Kroon Assistant Manager, Nuclear Magnetic Resonance Facilities Michael E. Pique Director, Graphics Development Nahid Razi, Ph.D. Assistant Core Manager, Consortium for Functional Glycomics Peter Sobieszcsuk, Ph.D. Core Manager, Consortium for Functional Glycomics Ryan Burnett, Ph.D. David Alvarez-Carbonell, Ph.D. Qing Chai, M.D., Ph.D. Jianghong An, Ph.D.** British Columbia Cancer Agency Vancouver, British Columbia Brian Chapados, Ph.D. Anju Chatterji, Ph.D. Adrienne Elizabeth Dubin, Ph.D. Yu An, Ph.D. Anton Vladislavovich Cheltsov, Ph.D. Maria Alejandra GamezAbascal, Ph.D. Crystal Stacy Anglen, Ph.D.** Neurome, Inc. La Jolla, California Jianhan Chen, Ph.D. Peter B. Hedlund, M.D., Ph.D. Brigitte Anliker, Ph.D. Yen-Ju Chen, Ph.D. Ying Chuan Lin, Ph.D. Roger Armen, Ph.D. Zhiyong Chen, Ph.D. Rebecca Page, Ph.D.** Brown University Providence, Rhode Island Joseph W. Arndt, Ph.D. Mabelle Ashe, Ph.D. Jaeyoung Cho, Ph.D.** Hallym University Kangwon, South Korea Mikhail Popkov, Ph.D. Jamie Mitchell Bacher, Ph.D. Jungwoo Choe, Ph.D. Richard R. Rivera, Ph.D. Chung Jen Chou, Ph.D. Jean-Pierre Clamme, Ph.D. S E N I O R S TA F F S C I E N T I S T Lincoln Scott, Ph.D. Michael F. Bailey, Ph.D.** Bio21 Institute Parkville, Victoria, Australia Wayne A. Fenton, Ph.D. Koji Tamura, Ph.D. Manidipa Banerjee, Ph.D. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Eli Chapman, Ph.D. Li-Chiou Chuang, Ph.D. MOLECULAR BIOLOGY 2005 157 Reza Mobini Farahani, Ph.D.** Sahlgrenska University Hospital Göteborg, Sweden Joy Huffman, Ph.D.** McKinsey & Company Los Angeles, California Shantanu Kumar, Ph.D. Daniel Felitsky, Ph.D. Laura M. Hunsicker, Ph.D.** Trinity University San Antonio, Texas Hugo Alfredo Lago-Zarrilli, Ph.D. †††† Allan Chris Merrera Ferreon, Ph.D. Kwan Hoon Hyun, Ph.D. Josephine Chu Ferreon, Ph.D. Wonpil Im, Ph.D. Sanjib Das, Ph.D. Pierre Henri Gaillard, Ph.D. Paramita Dasgupta, Ph.D.** Mayo Clinic Rochester, Minnesota Hui Gao, Ph.D. Tasneem Islam, Ph.D.** University of Melbourne Melbourne, Australia Robert De Bruin, Ph.D. Shannon E. Gardell, Ph.D. Shuichiro Ito, Ph.D.** Sankyo Co., Ltd. Tokyo, Japan Edith Caroline Glazer, Ph.D. Kai Jenssen, Ph.D. Bettina Groschel, Ph.D. Glenn C. Johns, Ph.D. Linda Maria Columbus, Ph.D. Adam Corper, Ph.D. Qizhi Cui, Ph.D. Carla P. Da Costa, Ph.D. Douglas Daniels, Ph.D.** Yale University New Haven, Connecticut Iaroslav Kuzmin, Ph.D. †††† Bianca Lam, Ph.D. Polo Chun Hung Lam, Ph.D. Emma Langley, Ph.D. Roberto N. De Guzman, Ph.D.** University of Kansas Lawrence, Kansas Sohela De Rozieres, Ph.D. Qingdong Deng, Ph.D. Paula Desplats, Ph.D. Buchi Ramachary Dhevalapally, Ph.D.** University of Hyderabad Hyderabad, India Claire Louise Dovey, Ph.D. Elsa D. Garcin, Ph.D. Björn Grünenfelder, Ph.D.** Novartis Institutes for BioMedical Research Cambridge, Massachusetts Fang Guo, Ph.D. Gye Won Han, Ph.D. Hongna Han, Ph.D.** American BioScience, Inc. Santa Monica, California Zhanna Druzina, Ph.D. Shoufa Han, Ph.D. Li-Lin Du, Ph.D. Wenge Han, Ph.D. Theresia Dunzendorfer-Matt, Ph.D.** Leopold Franzens Universität Innsbruck, Austria Jason W. Harger, Ph.D. Chul Won Lee, Ph.D. Jinhyuk Lee, Ph.D. Eric C. Johnson, Ph.D. June Hyung Lee, Ph.D. Margaret Alice Johnson, Ph.D. Kelly Lee, Ph.D. Hamid Reza Kalhor, Ph.D.†††† Katrina Lehmann, Ph.D. †††† Christian Kannemeier, Ph.D. Chenglong Li, Ph.D. Mili Kapoor, Ph.D. Vasco Liberal, Ph.D. Andrey Aleksandrovich Karyakin, Ph.D. William M. Lindstrom, Ph.D. Yang Khandogin, Ph.D. Hui-Yue Christine Lo, Ph.D. Ilja V. Khavrutskii, Ph.D. Kunheng Luo, Ph.D. Reza Khayat, Ph.D. John Gately Luz, Ph.D.** Harvard University Boston, Massachusetts David M. Herman, Ph.D. Deron Herr, Ph.D. Eda Koculi, Ph.D. Kenichi Hitomi, Ph.D. Milka Kostic, Ph.D. Reto Horst, Ph.D. Julio Kovacs, Ph.D. Laurent Magnenat, Ph.D.** Serono Pharmaceutical Research Institute SA Geneva, Switzerland Yunfeng Hu, Ph.D. Irina Kufareva, Ph.D. Darly Joseph Manayani, Ph.D. Ann MacLaren, Ph.D. Stephen Edgcomb, Ph.D. Susanna V. Ekholm-Reed, Ph.D. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Che Ma, Ph.D.** Academia Sinica Taipei, Taiwan Min Ju Kim, Ph.D.** Genomics Institute of the Novartis Research Foundation San Diego, California Scott Eberhardy, Ph.D. Marc-Olivier Ebert, Ph.D.** Leopold Franzens Universität Innsbruck, Austria Jonathan C. Lansing, Ph.D.** Momenta Pharmaceuticals Cambridge, Massachusetts Chang-Wook Lee, Ph.D. Dae Hee Kim, Ph.D. Brian Henriksen, Ph.D.** Eurogentec North America, Inc. San Diego, California Jason Lanman, Ph.D. 158 MOLECULAR BIOLOGY 2005 Jeff Mandell, Ph.D. Wataru Nomura, Ph.D. Stephanie Pond, Ph.D. Holly Heaslet Soutter, Ph.D. Maria Victoria MartinSanchez, Ph.D. Brian Nordin, Ph.D.** ActivX Biosciences, Inc. La Jolla, California Owen Pornillos, Ph.D. Natalie Spielewoy, Ph.D.** Weatherall Institute of Molecular Medicine Oxford, England Karin E. Norgard-Sumnicht, Ph.D.** San Diego State University San Diego, California Plachikkat Krishnan Radha, Ph.D. †††† Brian V. Norledge, Ph.D. John Reader, Ph.D.** University of North Carolina Chapel Hill, North Carolina Daniel Joseph Price, Ph.D. Tsutomu Matsui, Ph.D. Daniel McElheny, Ph.D.** University of Chicago Chicago, Illinois Benoit Melchior, Ph.D.** University of California Riverside, California David Metzgar, Ph.D.** Naval Health Research Center San Diego, California Jonathan Mikolosko, Ph.D. Susumu Mitsumori, Ph.D. Heiko Michael Moeller, Ph.D.** Universität Konstanz Konstanz, Germany Seongho Moon, Ph.D. Bettina Moser, Ph.D.** University of Illinois at Chicago Chicago, Illinois Samrat Mukhopadhyay, Ph.D. Christopher Myers, Ph.D.** Naval Health Research Center San Diego, California Sreenivasa Chowdari Naidu, Ph.D.** MediVas, L.L.C. San Diego, California Michael Oberhuber, Ph.D.** Leopold Franzens Universität Innsbruck, Austria Wendy Fernandez Ochoa, Ph.D. Grazia Daniela Raffa, Ph.D. Stevens Kastrup Rehen, Ph.D.** Universidade Federal do Rio de Janeiro Rio de Janeiro, Brazil Amy Odegard, Ph.D. Yoshiaki Zenmei Ohkubo, Ph.D.** Rutgers University Piscataway, New Jersey Brian L. Olson, Ph.D. Brian Paegel, Ph.D. Covadonga Paneda, Ph.D.** Molecular and Integrative Neurosciences Department, Scripps Research Sandeep Patel, Ph.D. Natasha Paul, Ph.D.** Stratagene, Inc. La Jolla, California Stephanie Pebernard, Ph.D. Jean-Baptiste Reiser, Ph.D.** European Synchrotron Radiation Facility Grenoble, France Miguel A. RodriguezGabriel, Ph.D.** Universidad Complutense de Madrid Madrid, Spain Stanislav Rudyak, Ph.D. Sean Ryder, Ph.D. Sanjay Adrian Saldanha, Ph.D. Sanjita Sasmal, Ph.D. †††† Greg Springsteen, Ph.D. Deborah J. Stauber, Ph.D.** Novartis Institutes for BioMedical Research Cambridge, Massachusetts Derek Steiner, Ph.D.** Johnson & Johnson San Diego, California Gudrun Stengel, Ph.D. Daniel Stoffler, Ph.D.** Universität Basel Basel, Switzerland Kenji Sugase, Ph.D. Vidyasankar Sundaresan, Ph.D.** GE Infrastructure Trevose, Pennsylvania Magnus Sundstrom, Ph.D. Jeff Suri, Ph.D.** GluMetrics, Inc. Long Beach, California Blair R. Szymczyna, Ph.D. Florence Muriel Tama, Ph.D. Mika Aoyagi Scharber, Ph.D.** Burnham Institute La Jolla, California Jinghua Tang, Ph.D.** University of California San Diego, California Toru M. Nakamura, Ph.D.** University of Illinois at Chicago Chicago, Illinois Suzanne Peterson, Ph.D.** University of California San Diego, California Jennifer S. Scorah, Ph.D. Nardos Tassew, Ph.D. Pedro Serrano-Navarro, Ph.D. Hiroaki Tateno, Ph.D. Sujatha Narayan, Ph.D. Craig McLean Shepherd, Ph.D. Hung Nguyen, Ph.D. Wolfgang Stefan Peti, Ph.D.** Brown University Providence, Rhode Island Michela Taufer, Ph.D.** University of Texas El Paso, Texas Tadateru Nishikawa, Ph.D. Goran Pljevaljcic, Ph.D. Eishi Noguchi, Ph.D.** Drexel University Philadelphia, Pennsylvania Corinne Chantal Ploix, Ph.D.** Novartis International AG Basel, Switzerland Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. William Shih, Ph.D** Dana Farber Cancer Institute Boston, Massachusetts David S. Shin, Ph.D. Develeena Shivakumar, Ph.D. Ewan Richardson Taylor, Ph.D. Donato Tedesco, Ph.D.** Berlex Biosciences Richmond, California MOLECULAR BIOLOGY 2005 159 Hua Tian, Ph.D. Lan Xu, Ph.D. Padmaja Natarajan, Ph.D. Rhonda Torres, Ph.D.** Merck & Co. Rahway, New jersey Yoshiki Yamada, Ph.D. Marianne Patch, Ph.D. Atsushi Yamagata, Ph.D. Gabriela Perez-Alvarado, Ph.D. Megan Wright Trevathan, Ph.D.** Harvard Medical School Boston, Massachusetts Qi Yan, Ph.D. Nicholas Preece, Ph.D. Yong Yao, Ph.D. Lin Wang, Ph.D. Xiaoqin Ye, M.D., Ph.D. Ulrich Ignaz Tschulena, Ph.D. Yongjun Ye, Ph.D. Julie L. Tubbs, Ph.D. VISITING I N V E S T I G AT O R S Yong Yin, Ph.D. Naoto Utsumi, Ph.D. Veronica Yu, Ph.D. Frank van Drogen, Ph.D. Stephen J. Benkovic, Ph.D. Pennsylvania State University University Park, Pennsylvania * Joint appointment in The Skaggs Institute for Chemical Biology ** Appointment completed; new location shown *** Joint appointment in the Molecular and Integrative Neurosciences Department **** Joint appointments in the Department of Immunology and The Skaggs Institute for Chemical Biology ***** Joint appointments in the Department of Chemistry and The Skaggs Institute for Chemical Biology † †† Yuan Yuan, Ph.D. Philip Arno Venter, Ph.D. Markus Zeeb, Ph.D. Petra Verdino, Ph.D. Astrid Graslund, Ph.D. Stockholm University Stockholm, Sweden Ying Zeng, Ph.D. Stefan Vetter, Ph.D.** Florida Atlantic University Boca Raton, Florida Haile Zhang, Ph.D. Arne Holmgren, M.D., Ph.D. Karolinska Institutet Stockholm, Sweden Yong Zhao, Ph.D. William Frederick Waas, Ph.D. Peizhi Zhu, Ph.D. Barry Honig, Ph.D. Columbia University New York, New York Shun-ichi Wada, Ph.D. S C I E N T I F I C A S S O C I AT E S Ross Walker, Ph.D. Enrique Abola, Ph.D. Arthur Horwich, M.D. Yale University New Haven, Connecticut Robert Scott Williams, Ph.D. Andrew S. Arvai, M.S. Raphaelle WinskySommerer, Ph.D.** Universität Zürich Zürich, Switzerland Eric Birgbauer, Ph.D. Ognian V. Bohorov, Ph.D. Eric L. Wise, Ph.D. Dennis Carlton, B.S. Jonathan Wojciak, Ph.D. Ellen Yu-Lin Tsai Chien, Ph.D. Dennis Wolan, Ph.D.** Sunesis Pharmaceuticals, Inc. South San Francisco, California Hyung Sik Won, Ph.D.** Konkuk University Chungju, Korea Xiaoping Dai, Ph.D. Liliane Dickinson, Ph.D. †††† Michael Allen Hanson, Ph.D. Diane Marie Kubitz, B.A. Timothy I. Wood, Ph.D. Marcy A. Kingsbury, Ph.D. Eugene Wu, Ph.D. Rolf Mueller, Ph.D. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Tai-huang Huang, Ph.D. Academica Sinica Taipei, Taiwan Robert D. Rosenstein, Ph.D. Lawrence Berkeley National Laboratory Berkeley, California Joint appointment in the Department of Cell Biology Joint appointment in the Department of Molecular and Experimental Medicine ††† Joint appointment in the Department of Chemistry †††† Appointment completed 160 MOLECULAR BIOLOGY 2005 Peter E. Wright, Ph.D. Chairman’s Overview esearch in the Department of Molecular Biology encompasses a broad range of disciplines, extending from structural and computational biology at one extreme to molecular genetics at the other. During the past year, our scientists continued to make rapid progress toward understanding the fundamental molecular events that underlie the processes of life. Major advances have been made in elucidating the structural biology of signal transduction and viral assembly, in understanding mechanisms of viral infectivity, in determining the structure of membrane proteins, in understanding the molecular basis of nucleic acid recognition and DNA repair, and in determining the mechanism of ribosome assembly. Progress was made in elucidating the molecular events involved in regulation of the cell cycle, in tumor development, in induction of sleep, in the molecular origins of neuronal development and of CNS disorders, in the regulation of transcription, and in the decoding of genetic information in translation. Finally, new advances were made in the design of novel low molecular weight compounds that can specifically regulate genes and in the area of biomolecular engineering, building novel functions into viruses, antibodies, zinc finger proteins, RNA, and DNA. Progress in these and other areas is described in detail on the following pages, and only a few highlights are mentioned here. R Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Structural biology continues to be a major activity in the department, and many new x-ray and nuclear magnetic resonance structures of major biomedical importance were completed during the past year. Among the highlights was the determination, in Ian Wilson’s laboratory, of the first structure of a human Toll-like receptor, a protein that plays a key role in the innate immune system as a sensor of molecules associated with the cell wall and genetic material of pathogenic bacteria. Dr. Wilson and his coworkers also reported structures of the protein CD1a, another key receptor in the innate immune response, and of an antibody that neutralizes most strains of HIV. Other advances came in the area of membrane protein crystallography: Geoffrey Chang and colleagues determined the structures of 2 proteins (MsbA and EmrE) involved in drug transport and the development of drug resistance in bacteria and cancer cells, and David Stout and James Fee determined the structure of a cytochrome ba 3 oxidase. Finally, the Joint Center for Structural Genomics, directed by Ian Wilson, was selected by the National Institutes of Health as 1 of 4 large-scale centers for high-throughput determination of protein structures. Several research groups are working in areas directly related to drug discovery and protein therapeutics. Joel Gottesfeld and colleagues have developed a small DNAbinding molecule that turns off the gene for histone H4 and blocks replication in a wide variety of cancer cells. The compound is active in vivo and blocks the growth of tumors in mice. Research in the laboratory of Carlos Barbas is directed toward genetic reprogramming of tumor cells via engineered zinc finger transcription factors. These artificial transcription factors are powerful tools for determining the function of genes in tumor growth and progression and have potential applications in cancer therapy. John Elder and colleagues are studying development of resistance to drugs that target the HIV protease. A complementary approach to the same problem is being taken by Arthur Olson and researchers in his laboratory in their FightAIDS@Home program. This program is a large-scale computational effort in which a grid of personal computers distributed around the world is used to design effective therapeutic agents that target the HIV protease. Raymond Stevens and coworkers have engineered a phenylalanine ammonia lyase enzyme as a potential injectable therapeutic agent for treating phenylketonuria. Finally, Paul Schimmel and colleagues have identified a naturally occurring fragment of tryptophanyl-tRNA synthetase that is highly potent in arresting angiogenesis and is being introduced in a clinical setting for treatment of macular degeneration. MOLECULAR BIOLOGY 2005 Many of the research groups in the department are applying the tools of molecular genetics to understand the molecular basis of human disease. Jerold Chun and his colleagues recently established a relationship between lysophospholipid signaling and neuropathic pain. In addition, they made the surprising discovery that lysophosphatidic acid receptors play an important role in embryonic implantation and thereby influence female fertility. Research in the laboratory of Luis de Lecea has indicated that a newly discovered neuropeptide, neuropeptide S, plays a functional role in modulation of sleep and suppression of anxiety. Work in the laboratory of James Paulson has led to the development of novel microarray technology for profiling glycoproteins, a technology that could eventually be developed into a powerful diagnostic screen for various infections and diseases. On the more fundamental side, major advances have been made in understanding mechanisms of protein and RNA folding, both in vitro and in a cellular environment. Research in the laboratory of Martha Fedor has resulted in new insights into mechanisms by which RNA folds into its specific functional structures and has provided evidence that RNA chaperones mediate folding pathways in the cell. Work by James Williamson and colleagues has led to a detailed map of the assembly landscape of the 30S ribosome, providing new understanding of the mechanism by which assembly proceeds through a succession of RNA conformational changes and protein binding events. Arthur Horwich and coworkers have made major progress in elucidating the mechanism by which the chaperone ClpA mediates unfolding and translocation of proteins. Molecular biology remains a field of enormous opportunity and excitement. The scientists in the department are taking full advantage of powerful new technologies to advance our understanding of fundamental biological processes at the molecular level. Their discoveries will ultimately be translated into new advances in biotechnology and in medicine. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 161 162 MOLECULAR BIOLOGY 2005 INVESTIGATORS’ R EPORTS Structural Biology of Immune Recognition, Molecular Assemblies, and Anticancer Targets I.A. Wilson, R.L. Stanfield, J. Stevens, X. Zhu, Y. An, K. Beis, T.A. Bowden, D.A. Calarese, R.M.F. Cardoso, P.J. Carney, J.-W. Choe, A.L. Corper, M.D.M. Crispin, T.A. Cross, X. Dai, W.L. Densley, E.W. Debler, M.-A. Elsliger, S. Ferguson, G.W. Han, P.A. Horton, S. Ito, M.J. JimenezDalmaroni, M.S. Kelker, J.G. Luz, J.B. Reiser, E.B. Shillington, D.A. Shore, D.J. Stauber, R.S. Stefanko, J.A. Vanhnasy, P. Verdino, E. Wise, D.W. Wolan, L. Xu, M. Yu, D.M. Zajonc, Y. Zhang ur main research focus is concerned with macromolecules and molecular complexes related to the innate and adaptive immune responses, viral pathogenesis, protein trafficking, purine biosynthesis, and reproductive biology. We use x-ray crystallography to determine atomic structures of key proteins in these systems in order to interpret functional data to probe mechanisms and modes of interaction and to aid in the design of therapeutic agents as potential drugs or vaccines. O T H E I N N AT E I M M U N E S Y S T E M Toll-like receptors (TLRs) are important mammalian glycoproteins involved in innate immunity that recognize conserved structures in pathogens called pattern recognition motifs. We recently determined the 2.1-Å crystal structure of the extracellular domain of human TLR3, which is activated by double-stranded viral RNA. TLR3 forms a large horseshoelike structure with an outer diameter of 80 Å. Key features include a hydrophobic core formed by the conserved leucine-rich repeats and a continuous β-sheet that spans 270° of arc. We are also investigating other TLRs and their ligands to understand how microorganisms are initially sensed by the innate immune system. Our goal is to use the data to design novel selective agonists and antagonists of TLR signaling pathways. This research is being done in collaboration with R.J. Ulevitch and B. Beutler, Department of Immunology. Another family of pattern recognition molecules called peptidoglycan recognition proteins (PGRPs) interacts with peptidoglycans. We have determined the crystal structure of the “recognition” PGRP-SA at 1.56 Å. ComPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. parison of PGRP-SA with a “catalytic” PGRP-LB indicates overall structural conservation and a hydrophilic groove that most likely corresponds to the peptidoglycan core binding site. Approximately 22,500 intensive care patients across the United States die of septic shock syndrome every year. Recently, researchers found that a newly discovered receptor termed triggering receptor expressed on myeloid cells 1 (TREM-1) mediates septic shock. We determined structures of human and mouse TREM-1 immunoglobulin-type domains to 1.47 Å and 1.76 Å, respectively. These structural results provided insights into the nature of ligand recognition by the TREM family in innate immunity. The studies on TREMs and PGRPs are being done in collaboration with L. Teyton, Department of Immunology. CLASSICAL AND NONCLASSICAL MHC AND T-CELL RECEPTOR SIGNALING In cellular immunity, T-cell receptors (TCRs) sense invading pathogens by recognizing pathogen-derived peptide fragments presented by MHC molecules. The TCRs then act in concert with CD8 and CD3, which assist in transducing the antigen recognition signal. Aberrant signaling can result in numerous disease states. The αβ TCR coreceptor CD8 is an essential factor in the TCR-mediated activation of cytotoxic T lymphocytes. We are doing structural studies of the CD8αβ and the CD8αα isoforms and of other constituents of the TCR signaling complex. The CD1 family of nonclassical MHC molecules presents lipid antigens to CD1-restricted TCRs. Our recent crystal structure of mouse CD1d at 2.2 Å in complex with the exceptionally potent short-chain sphingolipid α-galactosyl ceramide (Fig. 1) reveals a precise hydro- F i g . 1 . The short-chain sphingolipid α-galactosyl ceramide bound to mouse CD1d. This sphingolipid is a strong agonist of natural killer T cells. Both alkyl chains of the ligand are buried deep inside the binding groove, whereas the galactose headgroup is optimally positioned on top of the binding groove to directly interact with the TCR. MOLECULAR BIOLOGY 2005 163 gen-bonding network that positions the galactose moiety. Other CD1 structures determined include those of CD1a with a bound sulfatide and with a lipopeptide that have revealed how dual- and single-chain lipids interact with the same CD1 molecule. Collaborators in this research include D.B. Moody and M.B. Brenner, Harvard Medical School, Boston, Massachusetts; C.-H. Wong, Department of Chemistry; L. Teyton, Department of Immunology; M. Kronenberg, La Jolla Institute for Allergy and Immunology, San Diego, California; V. Kumar, Torrey Pines Institute for Molecular Studies, San Diego, California; and Wayne Severn, AgResearch, Upper Hut, New Zealand. 1918 INFLUENZA VIRUS Flu is a contagious respiratory disease caused by influenza viruses. Of all the known pandemics in the history of humans, the 1918 influenza outbreak was the most destructive; according to estimates, 40 million persons died. As a member of the “flu consortium” funded by the National Institutes of Health, we are working toward a molecular understanding of why this particular influenza virus was so pathogenic and how it managed to evade the immune system so effectively. We have determined the structure of the hemagglutinin of the 1918 virus, and now we are investigating the other viral proteins. We recently analyzed the receptor specificity of the 1918 hemagglutinin by comparing its binding to a panel of carbohydrates with the binding of more modern human and avian viruses (Fig. 2). For these studies, we are using novel glycan array technology developed by O. Blixt and J. Paulson, Consortium for Functional Glycomics, La Jolla, California. F i g . 2 . Results for carbohydrate array binding of the 2 natural hemagglutinins from the influenza virus that circulated during the 1918 pandemic. Human-adapted viruses preferentially bind to receptors with a terminal sialic acid linked by an α2,6 linkage to a vicinal galactose, whereas avian-adapted viruses recognize an α2,3 linkage. Glycan array results are shown for 18SC (A/South Carolina/1/18; A), and 18NY (A/New York/1/18; B). These 2 hemagglutinins differ by a single point mutation that is sufficient to alter the carbohydrate specificity from exclusively α2,6 to mixed α2,6/α2,3. AGP indicates α1acid glycoprotein. HIV TYPE 1 NEUTRALIZING ANTIBODIES A vaccine effective against the HIV type 1 must elicit antibodies that neutralize all circulating strains of the virus. However, antibodies with such properties are extremely rare; to date, only a handful have been isolated. Crystal structures for 4 of these rare, potent, broadly neutralizing antibodies (b12, 2G12, 4E10, 447-52D) in complex with their viral antigens have revealed the structural basis for the effectiveness of the antibodies (Fig. 3). Our goal is to design compounds on the basis of this structural information (retrovaccinology) for testing as potential vaccines. The research on HIV is being done in collaboration with D. Burton, Department of Immunology; P. Dawson, Department of Cell Biology; C.-H. Wong, Department of Chemistry; S. Danishefsky, Sloan-Kettering Institute, New York, New York; J.K. Scott, Simon Fraser University, Burnaby, British Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 3 . Antigen binding site of the Fab fragment of 4E10, an antibody to gp41. 4E10 cross-reacts with more viral isolates (clades) than any other known HIV type 1 neutralizing antibody. The crystal structure of Fab 4E10 is shown in complex with a synthetic peptide that encompasses the highly conserved 4E10 epitope. The peptide (ball and stick) binds to the surface of Fab 4E10 (solid surface) in a shallow hydrophobic cavity in a helical conformation. The structure also suggests that the complementarity-determining region H3 loop of 4E10 may contact the cell membrane, because the loop is adjacent to the membrane-proximal epitope. 164 MOLECULAR BIOLOGY 2005 Columbia; S. Zolla-Pazner, New York University School of Medicine, New York, New York; J. Moore, Cornell University, Ithaca, New York; Repligen Corporation, Waltham, Massachusetts; H. Katinger, R. Kunert, and G. Stiegler, University für Bodenkultur, Vienna, Austria; and R. Wyatt and P. Kwong, Vaccine Research Center, National Institutes of Health, Bethesda, Maryland. PRIMITIVE IMMUNOGLOBULINS Cartilaginous fish are the phylogenetically oldest living organisms known to have components of the vertebrate adaptive immune system, such as antibodies, MHC molecules, and TCRs. Key to their immune response are heavy-chain, homodimeric immunoglobulins (“new antigen receptors” or IgNARs) in which the antigen-recognizing variable domains consist of only a single immunoglobulin domain. In collaboration with M. Flajnik, University of Maryland Medical School, Baltimore, Maryland, we determined the crystal structure for an IgNAR variable domain in complex with its lysozyme antigen (Fig. 4). The results revealed that 2 complementarity-determining regions are sufficient for antigen recognition. These and ongoing studies will determine whether the IgNAR variable domains are an evolutionary precursor to mammalian TCR and antibody immunoglobulin domains. reactions not catalyzed by naturally occurring enzymes. Examples currently under study include several cocainehydrolyzing antibodies that could act as possible therapeutic agents to counter cocaine overdose or addiction, highly efficient but widely acting aldolase antibodies, and antibodies that carry out proton abstraction from carbon (Fig. 5). The studies on catalytic antibodies are being done in collaboration with R.A. Lerner, C.F. Barbas, K.D. Janda, P.G. Schultz, F. Tanaka, P. Wentworth, and P. Wirsching, Department of Chemistry; D.W. Landry, Columbia University, New York, New York; and D. Hilvert, ETH Zürich, Zürich, Switzerland. C ATA LY T I C A N T I B O D I E S Catalytic antibodies can be generated to carry out many difficult and novel chemical reactions, including F i g . 5 . Antibody-combining site of 34E4 bound to hapten. Cata- lytic antibody 34E4 catalyzes the conversion of benzisoxazoles to salicylonitriles with high rates and multiple turnovers. This reaction is a widely used model system for studies of proton abstraction from carbon. The structure of 34E4 in complex with its hapten has revealed many similarities to biological counterparts that promote proton transfers. Nevertheless, the reliance of 34E4 on a single catalytic residue (GluH50) probably prevents it from achieving the rates of the most efficient enzymes. Two of the active-site water molecules are designated S1 and S21. The 3Fo-2Fc σA-weighted electron density map around the hapten and key active-site residues is contoured at 1.3 σ. Hydrogen bonds are shown as broken lines. Trp L91 forms a cation-π interaction with the guanidinium moiety of the hapten. F i g . 4 . Nurse shark IgNAR type I variable domain (tubes) bound EVOLUTION OF LIGAND RECOGNITION AND to its lysozyme antigen (solid surface). The IgNAR variable domains have an unusual antigen-binding site that contains only 2 of the 3 conventional complementarity-determining regions (CDRs), but it still binds antigen with nanomolar affinity via an interface comparable in size to conventional antibodies. Two other regions, HV2 and HV4, are also somatically mutated, suggesting that they may also be involved in antigen recognition for other IgNAR-antigen complexes. SPECIFICITY Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. The antibodies 1E9 and DB3 share a human germline precursor but recognize different ligands. Residues in the Diels-Alderase antibody 1E9 active site have been sequentially mutated by D. Hilvert to change the specificity of 1E9 to that of the steroid-binding DB3. MOLECULAR BIOLOGY 2005 165 Only 2 key residues in 1E9 are required to switch between the catalytic antibody activity and steroid binding that is 14,000-fold higher than in the original 1E9 antibody. Crystal structures of these steroid-bound 1E9 mutants show that although 1E9 and DB3 share similar steroid-binding properties, they surprisingly accomplish binding and specificity in a structurally distinct manner. BLUE AND PURPLE FLUORESCENT ANTIBODIES Antibodies generated against trans-stilbene have an interesting, unexpected photochemistry when bound to that hapten. Several of these antibodies bind stilbene with high affinity, yet have significantly different spectroscopic properties. Crystal structures have now been determined to probe the antibodies’ mechanism of action, and further biophysical and biochemical studies are being performed in the laboratories of our collaborators, R.A. Lerner, Department of Molecular Biology; K.D. Janda and F.E. Romesberg, Department of Chemistry; and H.G. Gray, California Institute of Technology, Pasadena, California. PROTEIN TRAFFICKING The Rab family GTPases are ubiquitously involved in regulation of membrane docking and fusion in endocytic and exocytic pathways. The tethering factor p115 is recruited by Rab1 to vesicles of coat protein complex II during budding from the endoplasmic reticulum and subsequently interacts with a set of SNARE proteins associated with the vesicles to promote targeting to the Golgi complex. In collaboration with W.E. Balch, Department of Cell Biology, we determined the crystal structure of p115 at 2.0 Å and localized the binding site on p115 for Rab1 by mutational analysis. E N Z Y M AT I C C A N C E R TA R G E T S The de novo purine biosynthesis pathway is the primary provider of purine nucleotides, which are converted to DNA building blocks. This biosynthesis pathway is a validated target for the development of anticancer drugs because of heavy dependence on it by fast-growing cells, such as tumor cells. We have focused on 2 folate-dependent enzymes in the pathway: glycinamide ribonucleotide transformylase and the bifunctional aminoimidazole carboxamide ribonucleotide transformylase inosine monophosphate cyclohydrolase (ATIC, Fig. 6). Crystal structures of these 2 enzymes in complex with many different classes of inhibitors have provided a valuable platform for development of antineoplastic agents. These investigations are being done in collaboration with D.L. Boger, Department of Chemistry; A.J. Olson, Department of Molecular Biology; G.P. Beardsley, Yale UniverPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 6 . The active site of ATIC in complex with a novel nonfolate inhibitor identified by virtual ligand screening. The inhibitor is depicted in ball-and-stick representation and is surrounded by 2Fo-Fc electron density contoured at 1σ. sity, New Haven, Connecticut; and S.J. Benkovic, Pennsylvania State University, University Park, Pennsylvania. GHMP KINASES IN REPRODUCTIVE BIOLOGY XOL-1 is the primary sex-determining signal from Caenorhabditis elegans. The crystal structure of XOL-1 revealed that the protein belongs to the GHMP kinase family of small-molecule kinases, establishing an unanticipated role for this protein family in differentiation and development. In collaboration with B.J. Meyer, University of California, Berkeley, California, we identified XOL-1 homologs in the genomes of Caenorhabditis briggsae and Caenorhabditis remanei and are examining their function by using suppression of gene expression mediated by RNA interference. Although XOL-1 is structurally similar to its GHMP kinase neighbors, its endogenous ligand is unknown. Using the crystal structure of XOL-1 as a template for virtual screening, we identified several potential synthetic XOL-1 ligands, and in collaboration with J.R. Williamson, Department of Molecular Biology, we confirmed their binding by using nuclear magnetic resonance. JOINT CENTER FOR STRUCTURAL GENOMICS The Joint Center for Structural Genomics is a large consortium of scientists from Scripps Research, the Stanford Synchrotron Radiation Laboratory, the University of California, San Diego, the Burnham Institute, and the Genomics Institute of the Novartis Research Foundation. The center is funded by the Protein Structure 166 MOLECULAR BIOLOGY 2005 Initiative of the National Institute of General Medical Sciences. Its purpose is the high-throughput structure determination of the complete proteomes of a procaryote, Thermotoga maritima, and a eukaryote, the mouse. To date, members of the consortium have pioneered the development of many novel high-throughput methods, constructed a high-throughput pipeline, and determined more than 200 nonredundant structures, including 100 in the past year. PUBLICATIONS Arndt, J.W., Schwarzenbacher, R., Page, R., et al. Crystal structure of an / serine hydrolase (YDR428C) from Saccharomyces cerevisiae at 1.85 Å resolution. Proteins 58:755, 2005. Bakolitsa, C., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of an orphan protein (TM0875) from Thermotoga maritima at 2.00-Å resolution reveals a new fold. Proteins 56:607, 2004. Blixt, O., Head, S., Mondala, T., Scanlan, C., Huflejt, M.E., Alvarez, R., Bryan, M.C., Fazio, F., Calarese, D., Stevens, J., Razi, N., Stevens, D.J., Skehel, J.J., van Die, I., Burton, D.R., Wilson, I.A., Cummings, R., Bovin, N., Wong, C.H., Paulson, J.C. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. U. S. A. 101:17033, 2004. Bryan, M.C., Fazio, F., Lee, H.K., Huang, C.Y., Chang, A., Best, M.D., Calarese, D.A., Blixt, O., Paulson, J.C., Burton, D., Wilson, I.A., Wong, C.-H. Covalent display of oligosaccharide arrays in microtiter plates. J. Am. Chem. Soc. 126:8640, 2004. Canaves, J.M., Page, R., Wilson, I.A., Stevens, R.C. Protein biophysical properties that correlate with crystallization success in Thermotoga maritima: maximum clustering strategy for structural genomics. J. Mol. Biol. 344:977, 2004. Cardoso, R.M., Zwick, M.B., Stanfield, R.L., Kunert, R., Binley, J.M., Katinger, H., Burton, D.R., Wilson, I.A. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22:163, 2005. Crispin, M.D., Ritchie, G.E., Critchley, A.J., Morgan, B.P., Wilson, I.A., Dwek, R.A., Sim, R.B., Rudd, P.M. Monoglucosylated glycans in the secreted human complement component C3: implications for protein biosynthesis and structure. FEBS Lett. 566:270, 2004. Debler, E.W., Ito, S., Seebeck, F.P., Heine, A., Hilvert, D., Wilson, I.A. Structural origins of efficient proton abstraction from carbon by a catalytic antibody. Proc. Natl. Acad. Sci. U. S. A. 102:4984, 2005. Foss, T.R., Kelker, M.S., Wiseman, R.L., Wilson, I.A., Kelly, J.W. Kinetic stabilization of the native state by protein engineering: implications for inhibition of transthyretin amyloidogenesis. J. Mol. Biol. 347:841, 2005. Han, G.W., Schwarzenbacher, R., Page, R., et al. Crystal structure of an alanineglyoxylate aminotransferase from Anabaena sp at 1.70 Å resolution reveals a noncovalently linked PLP cofactor. Proteins 58:971, 2005. Hava, D.L., Brigl, M., van den Elzen, P., Zajonc, D.M., Wilson, I.A., Brenner, M.B. CD1 assembly and the formation of CD1-antigen complexes. Curr. Opin. Immunol. 17:88, 2005. Heine, A., Canaves, J.M., von Delft, F., et al. Crystal structure of O-acetylserine sulfhydrylase (TM0665) from Thermotoga maritima at 1.8 Å resolution. Proteins 56:387, 2004. Kelker, M.S., Debler, E.W., Wilson, I.A. Crystal structure of mouse triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.76 Å. J. Mol. Biol. 344:1175, 2004. Kelker, M.S., Foss, T.R., Peti, W., Teyton, L., Kelly, J.W., Wüthrich, K., Wilson, I.A. Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47 Å. J. Mol. Biol. 342:1237, 2004. Larsen, N.A., de Prada, P., Deng, S.X., Mittal, A., Braskett, M., Zhu, X., Wilson, I.A., Landry, D.W. Crystallographic and biochemical analysis of cocaine-degrading antibody 15A10. Biochemistry 43:8067, 2004. Levin, I., Miller, M.D., Schwarzenbacher, R., et al. Crystal structure of an indigoidine synthase A (IndA)-like protein (TM1464) from Thermotoga maritima at 1.90 Å resolution reveals a new fold. Proteins 59:864, 2005. Levin, I., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of a putative NADPH-dependent oxidoreductase (GI: 18204011) from mouse at 2.10 Å resolution. Proteins 56:629, 2004. Levin, I., Schwarzenbacher, R., Page, R., et al. Crystal structure of a PIN (PilT N-terminus) domain (AF0591) from Archaeoglobus fulgidus at 1.90 Å resolution. Proteins 56:404, 2004. Li, C., Xu, L., Wolan, D.W., Wilson, I.A., Olson, A.J. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J. Med. Chem. 47:6681, 2004. Mathews, I., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) from Thermotoga maritima at 2.0 Å resolution reveals a new fold. Proteins 59:869, 2005. McMullan, D., Schwarzenbacher, R., Hodgson, K.O., et al. Crystal structure of a novel Thermotoga maritima enzyme (TM1112) from the cupin family at 1.83 Å resolution. Proteins 56:615, 2004. Miller, M.D., Schwarzenbacher, R., von Delft, F., et al. Crystal structure of a tandem cystathionine-β-synthase (CBS) domain protein (TM0935) from Thermotoga maritima at 1.87 Å resolution. Proteins 57:213, 2004. Page, R., Peti, W., Wilson, I.A., Stevens, R.C., Wüthrich, K. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc. Natl. Acad. Sci. U. S. A. 102:1901, 2005. Pantophlet, R., Wilson, I.A., Burton, D.R. Improved design of an antigen with enhanced specificity for the broadly HIV-neutralizing antibody b12. Protein Eng. Des. Sel. 17:749, 2004. Reiser, J.B., Teyton, L., Wilson, I.A. Crystal structure of the Drosophila peptidoglycan recognition protein (PGRP)-SA at 1.56 Å resolution. J. Mol. Biol. 340:909, 2004. Santelli, E., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of a glycerophosphodiester phosphodiesterase (GDPD) from Thermotoga maritima (TM1621) at 1.60 Å resolution. Proteins 56:167, 2004. Schwarzenbacher, R., Jaroszewski, L., von Delft, F., et al. Crystal structure of an aspartate aminotransferase (TM1255) from Thermotoga maritima at 1.90 Å resolution. Proteins 55:759, 2004. Schwarzenbacher, R., Jaroszewski, L., von Delft, F., et al. Crystal structure of a type II quinolic acid phosphoribosyltransferase (TM1645) from Thermotoga maritima at 2.50 Å resolution. Proteins 55:768, 2004. Schwarzenbacher, R., von Delft, F., Jaroszewski, L., et al. Crystal structure of a putative oxalate decarboxylase (TM1287) from Thermotoga maritima at 1.95 Å resolution. Proteins 56:392, 2004. Heine, A., Luz, J.G., Wong, C.H., Wilson, I.A. Analysis of the class I aldolase binding site architecture based on the crystal structure of 2-deoxyribose-5-phosphate aldolase at 0.99 Å resolution. J. Mol. Biol. 343:1019, 2004. Spraggon, G., Pantazatos, D., Klock, H.E., Wilson, I.A., Woods, V.L., Jr., Lesley, S.A. On the use of DXMS to produce more crystallizable proteins: structures of the T maritima proteins TM0160 and TM1171 [published correction appears in Protein Sci. 14:1688, 2005]. Protein Sci. 13:3187, 2004. Jaroszewski, L., Schwarzenbacher, R., von Delft, F., et al. Crystal structure of a novel manganese-containing cupin (TM1459) from Thermotoga maritima at 1.65 Å resolution. Proteins 56:611, 2004. Spraggon, G., Schwarzenbacher, R., Kreusch, A., et al. Crystal structure of a methionine aminopeptidase (TM1478) from Thermotoga maritima at 1.9 Å resolution. Proteins 56:396, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. MOLECULAR BIOLOGY 2005 167 Spraggon, G., Schwarzenbacher, R., Kreusch, A., et al. Crystal structure of a Udpn-acetylmuramate-alanine ligase MurC (TM0231) from Thermotoga maritima at 2.3 Å resolution. Proteins 55:1078, 2004. Stanfield, R.L., Dooley, H., Flajnik, M.F., Wilson, I.A. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science 305:1770, 2004. Wang, X., Matteson, J., An, Y., Moyer, B., Yoo, J.S., Bannykh, S., Wilson, I.A., Riordan, J.R., Balch, W.E. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 167:65, 2004. Xu, L., Li, C., Olson, A.J., Wilson, I.A. Crystal structure of avian aminoimidazole4-carboxamide ribonucleotide transformylase in complex with a novel non-folate inhibitor identified by virtual ligand screening. J. Biol. Chem. 279:50555, 2004. Xu, Q., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of a formiminotetrahydrofolate cyclodeaminase (TM1560) from Thermotoga maritima at 2.80 Å resolution reveals a new fold. Proteins 58:976, 2005. Xu, Q., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of a ribose-5phosphate isomerase RpiB (TM1080) from Thermotoga maritima at 1.90 Å resolution. Proteins 56:171, 2004. Xu, Q., Schwarzenbacher, R., Page, R., et al. Crystal structure of an allantoicase (YIR029W) from Saccharomyces cerevisiae at 2.4 Å resolution. Proteins 56:619, 2004. Zajonc, D.M., Crispin, M.D., Bowden, T.A., Young, D.C., Cheng, T.Y., Hu, J., Costello, C.E., Rudd, P.M., Dwek, R.A., Miller, M.J., Brenner, M.B., Moody, D.B., Wilson, I.A. Molecular mechanism of lipopeptide presentation by CD1a. Immunity 22:209, 2005. Zhu, X., Tanaka, F., Hu, Y., Heine, A., Fuller, R., Zhong, G., Olson, A.J., Lerner, R.A., Barbas, C.F. III, Wilson, I.A. The origin of enantioselectivity in aldolase antibodies: crystal structure, site-directed mutagenesis, and computational analysis. J. Mol. Biol. 343:1269, 2004. Structure and Function of Proteins as Molecular Machines E.D. Getzoff, M. Aoyagi, A.S. Arvai, D.P. Barondeau, R.M. Brudler, T. Cross, E.D. Garcin, C. Hitomi, K. Hitomi, L. Holden, C.J. Kassmann, I. Li, M.E. Pique, M.E. Stroupe, J.L. Tubbs, T.I. Wood ur goals are to understand how proteins function as molecular machines. We use structural, molecular, and computational biology to study proteins of biological and biomedical interest, especially proteins that work synergistically with coupled chromophores, metal ions, or other cofactors. O PHOTOACTIVE PROTEINS AND CIRCADIAN CLOCKS To understand in atomic detail how proteins translate sunlight into defined conformational changes for biological functions, we are exploring the reaction mechanisms of the blue-light receptors photoactive yellow protein (PYP), photolyase, and cryptochrome. PYP is the prototype for the Per-Arnt-Sim domain proteins of circadian clocks, whereas proteins of the photolyase and cryptochrome family catalyze DNA repair or act in circadian clocks. To understand the protein photocycle (Fig. 1), we combined our ultra-high-resolution and Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 1 . Changes in the flexibility and mobility of PYP during its light cycle revealed by mapping the results of hydrogen-deuterium exchange mass spectrometry analyses (gray-scale shading) onto the x-ray crystallographic structure (ribbon showing overall protein fold). In the signaling state, regions of the protein including the N terminus are released for protein-protein interactions. time-resolved crystallographic structures of the dark state and 2 photocycle intermediates of PYP with sitedirected mutagenesis; ultraviolet-visible spectroscopy; time-resolved Fourier transform infrared spectroscopy; deuterium hydrogen exchange mass spectrometry, in collaboration with V. Woods, University of California, San Diego; and quantum mechanical and electrostatic computational methods, in collaboration with L. Noodleman, Department of Molecular Biology. Cryptochrome flavoproteins are homologs of lightdependent DNA repair photolyases that function as blue-light receptors in plants and as components of circadian clocks in animals. We determined the first crystallographic structure of a cryptochrome, which revealed commonalities with photolyases in DNA binding and redox-dependent function but showed differences in active-site and interaction surface features. New structures of photolyases from 2 other branches of the photolyase/cryptochrome family that repair cyclobutane pyrimidine dimers and photoproducts helped us decipher the cryptic structure, function, and evolutionary relationships of these fascinating redox-active proteins. A simple, but functional, circadian clock can be reconstituted in vitro from the 3 cyanobacterial proteins KaiA, KaiB, and KaiC alone. Yet, the structure and dynamics of the functional assembly of these proteins are not understood. Our crystallographic, dynamical light scattering and small-angle x-ray scattering studies revealed that KaiB self-assembles into a tetramer (Fig. 2). We are also studying clock proteins with PYP-like and Per-Arnt-Sim domains that bind to mammalian cryptochromes. Our goal is to determine the detailed chemistry and atomic structure of these pro- 168 MOLECULAR BIOLOGY 2005 F i g . 2 . The tetrameric assembly of the cyanobacterial circadian clock protein KaiB revealed by small-angle x-ray scattering (experimentally determined shape) and x-ray crystallography (ribbon showing protein fold). teins, define their mechanisms of action and interaction, and use our results to understand and regulate biological function. M E TA L L O E N Z Y M E S T R U C T U R E A N D F U N C T I O N Superoxide dismutases (SODs) act as master regulators of intracellular free radicals and reactive oxygen species by transforming superoxide to oxygen and hydrogen peroxide. Novel nickel SODs assemble into hollow spheres composed of six 4-helix bundle subunits. The 9 N-terminal residues fold into a unique nickel hook motif that shows promise as a detectable metal ion–binding tag in protein purification and structure determination. Our crystallographic structures of classic copper-zinc SODs from mammals, bacterial symbionts, and pathogens revealed striking differences in the enzyme assembly and in the loops flanking the active-site channel, despite the shared β-barrel subunit fold, catalytic metal center, and electrostatic enhancement of activity. With J. Tainer, Department of Molecular Biology, we determined structures of mutant human SODs found in patients with the disease amyotrophic lateral sclerosis (Lou Gehrig disease), and proposed a hypothesis for how single-site mutations cause this fatal neurodegenerative disease. To synthesize nitric oxide, a cellular signal and defensive cytotoxin, nitric oxide synthases (NOSs) require calmodulin-orchestrated interactions between their catalytic, heme-containing oxygenase module and their electronsupplying reductase module. Crystallographic structures Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. of wild-type and mutant NOS oxygenase dimers with substrate, intermediate, inhibitors, cofactors, and cofactor analogs, determined in collaboration with D. Stuehr, the Cleveland Clinic, Cleveland, Ohio, and J. Tainer, provided insights into the catalytic mechanism and dimer stability. Our structure-based drug design projects are aimed at selectively inhibiting inducible NOS, to prevent inflammatory disorders, or neuronal NOS, to prevent migraines, while maintaining blood pressure regulation by endothelial NOS. We integrated biochemical data with our structures of NOS oxygenase, NOS reductase, and calmodulin in complex with peptides derived from NOS to propose a model for the assembled holoenzyme that provides a moving-domain mechanism for electron flow from NADPH through 2 flavin cofactors to the heme. Our structure of the NOS reductase provides new insights into the complex regulatory mechanisms of this enzyme family. M E TA L L O P R O T E I N D E S I G N An ultimate goal for protein engineers is to design and construct new protein variants with desirable catalytic or physical properties. As members of the Scripps Research Metalloprotein Structure and Design Group, we are testing our understanding of the affinity, selectivity, and activity of metal ions by transplanting metal sites from structurally characterized metalloproteins into new protein scaffolds. To aid our design efforts, we have organized quantitative information and interactive viewing of protein metal sites at the Metalloprotein Database and Browser (available at http://metallo.scripps.edu). For green fluorescent protein and the homologous red fluorescent protein, we designed, constructed, and characterized metal-ion biosensors in which binding of metal ions is signaled by changes in the spectroscopic properties of the naturally occurring fluorophores. The green fluorescent protein scaffold provides advantages over existing probes by allowing optimization with random mutagenesis, noninvasive expression in living cells, and targeting to specific cellular locations. By completing the metalloprotein design cycle from prediction to highly accurate structures, we can rigorously evaluate and improve our algorithms for the design of metal sites. Our related structural studies of green and red fluorescent protein intermediates in chromophore cyclization and oxidation provide a novel mechanism for the spontaneous synthesis of these tripeptide fluorophores within the protein scaffold. MOLECULAR BIOLOGY 2005 PUBLICATIONS Barondeau, D.P., Getzoff, E.D. Structural insights into protein-metal ion partnerships. Curr. Opin. Struct. Biol. 14:765, 2004. Barondeau, D.P., Kassmann, C.J., Tainer, J.A., Getzoff, E.D. Understanding GFP chromophore biosynthesis: controlling backbone cyclization and modifying posttranslational chemistry. Biochemistry 44:1960, 2005. Dunn, A.R., Belliston-Bittner, W., Winkler, J.R., Getzoff, E.D., Stuehr, D.J., Gray, H.B. Luminescent ruthenium(II)- and rhenium(I)-diimine wires bind nitric oxide synthase. J. Am. Chem. Soc. 127:5169, 2005. Hitomi, K., Oyama, T., Han, S., Arvai, A.S., Getzoff, E.D. Tetrameric architecture of the circadian clock protein KaiB: a novel interface for intermolecular interactions and its impact on the circadian rhythm. J. Biol. Chem. 280:19127, 2005. Stroupe, M.E., Getzoff, E.D. The role of siroheme in sulfite and nitrite reductases. In: Tetrapyrroles: Their Birth, Life and Death. Warren, M.J., Smith, A. (Eds.). Landes Bioscience, Georgetown, Tex, in press. Stuehr, D.J., Wei, C.C., Santolini, J., Wang, Z., Aoyagi, M., Getzoff, E.D. Radical reactions of nitric oxide synthases. In: Free Radicals: Enzymology, Signaling, and Disease. Cooper, C.E., Wilson, M.T., Darley-Usmar, V.H. (Eds.). Portland Press, London, 2004, p. 39. Biochemical Society Symposia, Vol. 71. Tiso, M., Konas, D.W., Panda, K., Garcin, E.D., Sharma, M., Getzoff, E.D., Stuehr, D.J. C-terminal tail residue ARG1400 enables NADPH to regulate electron transfer in neuronal nitric oxide synthase. J. Biol. Chem., in press. Tubbs, J.L., Tainer, J.A., Getzoff, E.D. Crystallographic structures of Discosoma red fluorescent protein with immature and mature chromophores: linking peptide bond trans-cis isomerization and acylimine formation in chromophore maturation. Biochemistry 44:9833, 2005. Vevodova, J., Graham, R.M., Raux, E., Schubert, H.L., Roper, D.I., Brindley, A.A., Scott, A.I., Roessner, C.A., Stamford, N.P., Stroupe, M.E., Getzoff, E.D., Warren, M.J., Wilson, K.S. Structure/function studies on an S-adenosyl-L-methionine-dependent uroporphyrinogen III C methyltransferase (SUMT), a key regulatory enzyme of tetrapyrrole biosynthesis. J. Mol. Biol. 344:419, 2004. Wei, C.C., Wang, Z.Q., Durra, D., Hemann, C., Hille, R., Garcin, E.D., Getzoff, E.D., Stuehr, D.J. The three nitric-oxide synthases differ in their kinetics of tetrahydrobiopterin radical formation, heme-dioxy reduction, and arginine hydroxylation. J. Biol. Chem. 280:8929, 2005. Structural Molecular Biology of Interactions and Protein Design J.A. Tainer, A.S. Arvai, D.P. Barondeau, M. Bjoras, B.R. Chapados, L. Craig, T.H. Cross, D.S. Daniels, G. DiVita, L. Fan, C. Hitomi, K. Hitomi, J.L. Huffman, C.J. Kassmann, I. Li, G. Moncalian, M.E. Pique, D.S. Shin, O. Sundheim, R.S. Williams, T.I. Wood, A. Yamagata ur goals are to bridge the gap between the vastly improved tools and insights for structural cell biology at the molecular level and the applications of these advances for the molecular-based understanding of and eventual intervention in human diseases. Thus, our primary concern is the application of structural biology to fundamental questions of molecular and cellular biology relevant to human disease. Currently, we are investigating fundamental processes and principles of DNA repair, control of reactive oxygen species, control of the cell cycle, and pathogenesis. We think O Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 169 these processes have networked connections and common themes in terms of structural mechanisms and controls and medical implications. In general, our structural determination and design work involves hypothesis-driven studies; we focus on high-resolution structural analyses, functionally important conformational changes, and macromolecular interactions, including design of inhibitors and dynamic assemblies that act as macromolecular machines to control the fundamental processes of cell biology. To accomplish our basic research, we use protein crystallography, solution x-ray scattering, fluorescence, biochemistry, mutagenesis, and protein expression. Our experimental work is complemented by efforts to develop new methods, particularly in structural analysis, protein and drug design, and the merging of crystal structures with x-ray solution structures and electron microscopy. These new experimental integrations involve the use of synchrotron radiation to bridge the size and resolution gap between high-resolution macromolecular structures and the multiprotein macromolecular machines and reversible interactions in the cell. For protein design, we have an active collaboration with E. Getzoff, Department of Molecular Biology, to understand and control the formation of self-synthesizing chromophores in green fluorescent protein and its homologs. We are increasingly interested in structure-based design of inhibitors that are relevant to the development of novel therapeutic agents and inhibitors that chemically knock out or block gene function to complement genes that are knocked out by removing the DNA. The synergy between basic research and advances in techniques is allowing us to contribute to the basic understanding and treatment of degenerative and infectious diseases and cancer. S U P E R O X I D E D I S M U TA S E S Superoxide dismutases (SODs) are master regulators for reactive oxygen species involved in injury, pathogenesis, aging, and degenerative diseases. In basic research on these enzymes, we are characterizing the activity of the mitochondrial SODs. We discovered a novel nickel ion SOD and characterized its hexameric assembly. For the human cytoplasmic copper, zinc SOD, we examined how single-site mutations cause the neurodegenerative Lou Gehrig disease or familial amyotrophic lateral sclerosis (FALS). We found that point mutations destabilize the copper, zinc SOD dimer and dramatically increase its propensity to aggregate and form filaments that resemble those seen in motor neurons of patients with FALS. These findings provide a molecu- 170 MOLECULAR BIOLOGY 2005 lar basis for the notion that a single FALS disease phenotype arises from diverse point mutations throughout the protein that reduce the structural integrity of copper, zinc SOD and lower the energy barrier for fibrous aggregation. Additionally, our new high-resolution structures of a related thermophilic copper, zinc SOD showed a trapped product complex. This novel finding helps define the enzyme’s mechanism of action and its susceptibility to inactivation by hydrogen peroxide. DNA REPAIR All life requires constant repair of DNA. Structural and mutational analyses of DNA repair enzymes provide a framework for understanding the molecular basis of genetic integrity and the loss of this integrity in cancer and degenerative diseases. We are interested in how specific types of damage are detected, how repair enzymes are coordinated within different pathways, and the nature and role of conformational change in proteins and DNA in repair pathways. We use electron microscopy, x-ray crystallography, small-angle x-ray scattering, and complementary in vitro and in vivo mutational analysis to go from enzyme structures to repair pathways and the coordination of repair with replication and transcription. We focus on pathways for DNA base repair, DNA nick translation in repair and replication (Fig. 1), and repair of double-stranded breaks. Understanding the structural chemistry and cell biology of DNA repair is critical for designing specific inhibitors to increase the effectiveness of chemotherapy and also for assessing how DNA repair enzyme polymorphisms may affect diseases in humans. Currently, we are designing inhibitors of enzymes that repair alkylated and oxidized guanines. These enzymes are one of the body’s natural defenses against DNA damage, but they can also inadvertently protect cancer cells from chemotherapeutic agents. For example, the human repair protein O6-alkylguanine-DNA alkyltransferase, which acts in the repair of alkylated guanines, repairs damaged DNA inside human cells, and cancer cells can use it to repair DNA that has been damaged in the course of chemotherapy, thus making the chemotherapy ineffective. F i g . 1 . Interactions between the complex consisting of flap endo- nuclease 1 (FEN-1), DNA, and proliferating cell nuclear antigen (PCNA) and the interface of DNA repair and replication. A, Nicked DNA is protected and repaired by the sequential activities of DNA polymerase δ (pol δ) and FEN-1 held to DNA by the "sliding clamp" PCNA. In the absence of FEN-1, a complex of pol δ and PCNA binds to and protects the nick (top). FEN-1 initiates nick translation by binding to PCNA (bottom), recognizing the 3′ DNA flap and cleaving the 5′ flap, generating a nick translated by 1 nucleotide. B. Structures of FEN-1 bound to DNA show that FEN-1 recognizes the 3′ flap in a sequence-independent manner. C, A composite model of the FEN-1–DNA–PCNA complex suggests how a kinked DNA intermediate might facilitate sequential activities of FEN-1 and pol δ. assembly ATPase, the membrane anchor protein interactions, and the assembled pilus fiber (Fig. 2). Our electron BACTERIAL PILI Type IV pili are essential virulence factors for many gram-negative bacteria, playing key roles in surface motility, adhesion, formation of microcolonies and biofilms, natural transformation, and signaling. We have determined structures for the type IV pilin subunits and for the assembled pilus fiber. Currently, we are investigating the type IV pilus assembly system, including the Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . A schematic view of the assembly machinery of type IV pili: the electron cryomicroscopy structure of the pilus of Neisseria gonorrhoeae (GC); crystal structures of full-length Pseudomonas aeruginosa (P.a) pilin; BfpC, the binding partner protein to ATPase from enteropathogenic Escherichia coli; and GspE2, the hexameric assembly ATPase from Archaeoglobus fulgidus. MOLECULAR BIOLOGY 2005 microscopy and x-ray structures of protein components and complexes are helping us understand the architecture and assembly mechanism as a basis for the design of antibacterial vaccines and therapeutic agents. PUBLICATIONS Ayala, I., Perry, J.P., Szczepanski, J., Tainer, J.A., Vala, M.T., Nick, H.S., Silverman, D.N. Hydrogen bonding in human manganese superoxide dismutase containing 3-fluorotyrosine. Biophys. J., in press. Barondeau, D.P., Kassmann, C.J., Tainer, J.A., Getzoff, E.D. Understanding GFP chromophore biosynthesis: controlling backbone cyclization and modifying posttranslational chemistry. Biochemistry 44:1960, 2005. Crowther, L.J., Yamagata, A., Craig, L., Tainer, J.A., Donnenberg, M.S. The ATPase activity of BfpD is greatly enhanced by zinc and allosteric interactions with other Bfp proteins. J. Biol. Chem. 280:24839, 2005. de Jager, M., Trujillo, K.M., Sung, P., Hopfner, K.P., Carney, J.P., Tainer, J.A., Connelly, J.C., Leach, D.R., Kanaar, R., Wyman, C. Differential arrangements of conserved building blocks among homologs of the Rad50/Mre11 DNA repair protein complex. J. Mol. Biol. 339:937, 2004. Garcin, E.D., Bruns, C.M., Lloyd, S.J., Hosfield, D.J., Tiso, M., Gachhui, R., Stuehr, D.J., Tainer, J.A., Getzoff, E.D. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 279:37918, 2004. Hendrickson, E.A., Huffman, J.L., Tainer, J.A. Structural aspects of Ku and the DNA-dependent protein kinase complex. In: DNA Damage Recognition. Seide, W., Kow, Y.W., Doetsch, P.W. (Eds.). Taylor & Francis, New York, 2005, p. 629. Huffman, J.L., Sundheim, O., Tainer, J.A. DNA base damage recognition and removal: new twists and grooves. Mutat. Res. 577:55, 2005. Huffman, J.L., Sundheim, O., Tainer, J.A. Structural features of DNA glycosylases and AP endonucleases. In: DNA Damage Recognition. Seide, W., Kow, Y.W., Doetsch, P.W. (Eds.). Taylor & Francis, New York, 2005, p. 299. Manuel, R.C., Hitomi, K., Arvai, A.S., House, P.G., Kurtz, A.J., Dodson, M.L., McCullough, A.K., Tainer, J.A., Lloyd, R.S. Reaction intermediates in the catalytic mechanism of Escherichia coli MutY DNA glycosylase. J. Biol. Chem. 279:46930, 2004. Putnam, C.D.. Tainer, J.A. Protein mimicry of DNA and pathway regulation. DNA Repair (Amst.), in press. Sarker, A.H., Tsutakawa, S.E., Kostek, S., Ng, C., Shin, D.S., Peris, M., Campeau, E., Tainer, J.A., Nogales, E., Cooper, P.K. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne syndrome. Mol. Cell 20:187, 2005. Simeoni, F., Arvai, A., Bello, P., Gondeau, C., Hopfner, K.P., Neyroz, P., Heitz, F., Tainer, J., Divita, G. Biochemical characterization and crystal structure of a Dim1 family associated protein: Dim2. Biochemistry 44:11997, 2005. Tubbs, J.L., Tainer, J.A., Getzoff, E.D. Crystallographic structures of Discosoma red fluorescent protein with immature and mature chromophores: linking peptide bond trans-cis isomerization and acylimine formation in chromophore maturation. Biochemistry 44:9833, 2005. Williams, R.S., Tainer, J.A. A nanomachine for making ends meet: MRN is a flexing scaffold for the repair of DNA double-strand breaks. Mol. Cell 19:724, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 171 Structural Biology of Integral Membrane Proteins G. Chang, A. Chen, Y. Chen, X. He, O. Pornillos, C.R. Reyes, P. Szewczk, A. Ward, S. Wada, Y. Yin -ray crystallography of integral membrane proteins is an exciting and rapidly growing frontier in molecular structural biology. We are interested in 5 areas: (1) the molecular structural basis for lipid and drug transport across the cell membrane by multidrugresistance (MDR) transporters, (2) the high-resolution structure of yeast and mammalian MDR transporters, (3) signal transduction by receptors, (4) discovery and the structurally based design of potent MDR reversal agents, and (5) the development of an in vitro cell-free system capable of overproducing integral membrane proteins suitable for biophysical study. We use several experimental methods, including detergent/lipid protein biochemistry, 3-dimensional crystallization of integral membrane proteins, and x-ray crystallography. We are developing and using an efficient cell-free membrane protein expression system in collaboration with T. Kudlicki, Invitrogen Corporation, Carlsbad, California, for the overexpression integral membrane proteins for both x-ray crystallography and nuclear magnetic resonance studies. We are addressing the molecular basis of MDR, a significant challenge in the treatment of infectious disease and cancer. A major cause of MDR in both of these situations is a battery of drug efflux pumps imbedded in the cell membrane. Through our structural studies on MDR transporters, we hope to gain insights into the mechanics of translocating amphipathic substrates across the cell membrane and also the rational design of potent MDR reversal agents. We are combining chemistry and biology with structure for the discovery and design of potent MDR reversal agents for cancer chemotherapy in collaboration with M.G. Finn, Department of Chemistry; I. Urbatsch, Texas Tech University Health Sciences Center, Lubbock Texas; and S. Reutz, Novartis International AG, Basel, Switzerland. In collaboration with M. Saier, University of California, San Diego, and Q. Zhang, Department of Molecular Biology, we are determining the x-ray structures and mapping the detailed functional components of 3 families of bacterial MDR transporters that are dominant in gram-positive pathogens. In another collaboration, with R.A. Milligan, Department of Cell Biology, we are using electron cryomicroscopy to visualize the low-res- X 172 MOLECULAR BIOLOGY 2005 olution structures of our transporters. Through these united efforts, we will gain a broader understanding of the structure and function of drug transporters that cause MDR in cancer and bacterial infection. Recently, we determined a new structure of the MDR ATP-binding cassette transporter homolog MsbA in complex with magnesium, adenosine diphosphate, inorganic vanadate, and rough-chemotype lipopolysaccharide. This structure supports a model involving a rigid-body torque of the 2 transmembrane domains during ATP hydrolysis and suggests a mechanism by which the nucleotide-binding domain communicates with the transmembrane domain. We propose a lipid “flip-flop” mechanism in which the sugar groups are sequestered in the chamber while the hydrophobic tails are dragged through the lipid bilayer (Fig. 1). This posthydrolysis F i g . 1 . Proposed model for sequestering the polar sugar headgroup of lipopolysaccharide (LPS) in the internal chamber of MsbA (for clarity, only 1 LPS is shown). A, LPS initially binds to the elbow helix as modeled onto the closed apo structure. B, Lipid headgroups modeled to insert into the chamber of the apo closed structure. C, As the transporter undergoes conformational changes related to binding and hydrolysis of ATP, the headgroup is “flipped” within the polar chamber while the LPS hydrocarbon chains are freely exposed and dragged through the lipid bilayer. Both LPS and MsbA conforma- tions are modeled. D, LPS is presented to the outer leaflet of the membrane as observed in this structure. Reprinted with permission from Reyes, C.L., Chang, G. Science 308:1028, 2005. structure of MsbA also gives insight into the possible drug-binding sites for a number of cancer compounds. We are continuing our x-ray structural studies of the small MDR transporter EmrE and of other families of bacterial MDR transporters to better understand the molecular basis of the drug-proton antiport. The x-ray structures of MsbA and EmrE are excellent models for drug efflux systems that confer MDR to cancer cells and infectious microorganisms. PUBLICATIONS Ma, C., Chang, G. Crystallography of the integral membrane protein EmrE from Escherichia coli. Acta Crystallogr. D Biol. Crystallogr. 60:2399, 2004. Reyes, C.L., Chang, G. Structure of the ABC transporter MsbA in complex with ADP•vanadate and lipopolysaccharide. Science 308:1028, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Structure and Function of Membrane-Bound Enzymes C.D. Stout, H. Heaslet, M. Yamaguchi, V. Sundaresan, L. Hunsicker-Wang, J. Chartron ne focus of our research is the structure and function of transhydrogenase, an essential enzyme of respiration in mitochondria and bacteria. Transhydrogenase couples proton translocation across the membrane with hydride transfer between cofactors bound to soluble domains. We are determining the structure of the enzyme in its membrane-bound conformation and are studying the structures of the soluble domains. For studies of enzyme function, we are using biochemical methods and mutagenesis. Structural studies entail x-ray crystallography, electron microscopy studies done in collaboration with M. Yeager and B. Carragher, Department of Cell Biology, and nuclear magnetic resonance experiments done in collaboration with J. Dyson, Department of Molecular Biology. In collaboration with E.F. Johnson, Department of Molecular Biology, and J.R. Halpert, University of Texas Medical Branch, Galveston, Texas, we are studying highresolution crystal structures of mammalian cytochrome P450s. The P450s are monooxygenases involved in the biosynthesis and oxidation of lipophilic molecules, and they specifically metabolize a wide range of exogenous compounds and drugs. More than 60 genes for P450s occur in the human genome. We are studying high-resolution structures and drug-bound complexes of the human P450s 2C8, 2C9, 2A6, 3A4, and 1A2 and the rabbit P450s 2B4 and 2C5. In collaboration with J.A. Fee, Department of Molecular Biology, we are studying the structure and mechanism of cytochrome ba 3 oxidase, the terminal enzyme of respiration responsible for the reduction of molecular oxygen to water. The high-resolution crystal structure of the enzyme from a thermophilic bacterium has been determined (Fig. 1). Crystallographic experiments, in combination with mutagenesis and spectroscopy, are being used to capture intermediates in the reaction cycle and to define the pathways of proton translocation to and from the active site within the membrane. In parallel with these studies, we are developing the application of nanodiscs for biophysical studies of integral membrane proteins. These experiments are being done in collaboration with S.G. Sligar, University of Illinois, Urbana, Illinois, and M. Yeager, Department O MOLECULAR BIOLOGY 2005 173 Sundaresan, V., Chartron, J., Yamaguchi, M., Stout, C.D. Conformational diversity in NAD(H) and interacting transhydrogenase nicotinamide nucleotide binding domains. J. Mol. Biol. 346:617, 2005. Wester, M.R., Yano, J.K., Schoch, G.A., Yang, C., Griffin, K.J., Stout, C.D., Johnson, E.F. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-Å resolution. J. Biol. Chem. 279:35630, 2004. Yadav, M.K., Redman, J.E., Leman, L.J., Alvarez-Gutierrez, J.M., Zhang, Y., Stout, C.D., Ghadiri, M.R. Structure-based engineering of internal cavities in coiled-coil peptides. Biochemistry 44:9723, 2005. F i g . 1 . Crystal structure of the integral membrane protein cytochrome ba 3 oxidase from the thermophilic bacterium Thermus thermophilus. Cytochrome oxidase is responsible for the reduction of oxygen to water during respiration in all higher organisms. of Cell Biology. Nanodiscs are water-soluble particles that consist of 2 copies of an engineered construct of human apolipoprotein A-I (~200 amino acids) encircling a patch of bilayer containing the approximately 160 molecules of dimyristoyl-sn-glycero-3-phosphocholine or other phospholipids. Integral membrane proteins can be inserted into these particles by spontaneous self-assembly, and to date we have incorporated both cytochrome ba 3 oxidase and transhydrogenase. Additional research projects involve collaboration with other faculty members at Scripps Research. These projects include studies of iron-sulfur and electron transfer proteins, in collaboration with J.A. Fee and L. Noodleman, Department of Molecular Biology; RNA-protein complexes, with J.R. Williamson, Department of Molecular Biology; synthetic, self-assembling peptides, with M.R. Ghadiri, Department of Chemistry; and HIV protease inhibitor complexes, with A. Olson, Department of Molecular Biology, and B.E. Torbett, Department of Molecular and Experimental Medicine. PUBLICATIONS Carroll, K.S., Gao, H., Chen, H., Stout, C.D., Leary, J.A., Bertozzi, C.R. A conserved mechanism for sulfonucleotide reduction. PloS Biol. 3:e250, 2005. Fee, J.A., Todaro, T.R., Luna, E., Sanders, D., Hunsicker-Wang, L.M., Patel, K.M., Bren, K.L., Gomez-Moran, E., Hill, M.G., Ai, J., Loehr, T.M., Oertling, W.A., Williams, P.A., Stout, C.D., McRee, D., Pastuszyn, A. Cytochrome rC552, formed during expression of the truncated, Thermus thermophilus cytochrome c552 gene in the cytoplasm of Escherichia coli, reacts spontaneously to form protein-bound, 2-formyl-4-vinyl (Spirographis) heme. Biochemistry 43:12162, 2004. Hays, A.-M., Dunn, A.R., Chiu, R., Gray, H.B., Stout, C.D., Goodin, D.B. Conformational states of cytochrome P450cam revealed by trapping of synthetic wires. J. Mol. Biol. 344:455, 2004. Horne, W.S., Yadav, M.K., Stout, C.D., Ghadiri, M.R. Heterocyclic peptide backbone modifications in an α-helical coiled coil. J. Am. Chem. Soc. 126:15366, 2004. Hunsicker-Wang, L.M., Pacoma, R.L., Chen, Y., Fee, J.A., Stout, C.D. A novel cryoprotection scheme for enhancing diffraction of crystals of recombinant cytochrome ba3 oxidase from Thermus thermophilus. Acta Crystallogr. D Biol. Crystallogr. 61:340, 2005. Stout, C.D. Cytochrome P450 conformational diversity. Structure (Camb.) 12:1921, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Yano, J.K., Hsu, M.H., Griffin, K.J., Stout, C.D., Johnson, E.F. Structures of human microsomal cytochrome P450 2A6 complexed with coumarin and methoxsalen. Nat. Struct. Mol. Biol. 12:822, 2005. Yano, J.K., Wester, M.R., Schoch, G.A., Griffin, K.J., Stout, C.D., Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by x-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 279:38091, 2004. Lipid Chemistry for Studies of Integral Membrane Proteins Q. Zhang, M.G. Finn,* X. Ma * Department of Chemistry, Scripps Research ntegral membrane proteins float in the lipid bilayer with their hydrophobic domains threaded through the membrane and their hydrophilic domains extended into the aqueous solution. These proteins are extremely unstable outside the hydrophobic membrane bilayer, a situation that makes their in vitro biophysical and structural characterization difficult. An artificial environment is therefore needed to stabilize the proteins in their native state. We are attempting to synthesize new amphiphilic molecules that can extract integral membrane proteins from membranes and stabilize the proteins for structural characterization. Relatively few investigators have actually addressed questions about the design of appropriate amphiphilic molecules despite the extensive use of such molecules in studies of membrane proteins. The criteria that we apply to generate such amphiphilic molecules are based on the physical properties of the molecules and on their interactions with membrane proteins. Detergents that self-assemble into micellar structures are universally used to dissolve integral membrane proteins as single particles to facilitate protein crystallization. We intend to incorporate more hydrophobicity in the interior of detergent micelles to improve the stability of the micelles and consequently their ability to stabilize integral membrane proteins. We accomplish this incorporation by appending branches along the alkyl chains of detergents and, most interestingly, by adding a short branch at the interface between the hydrophobic tail and the hydro- I 174 MOLECULAR BIOLOGY 2005 philic head. These branches may behave in 2 distinct ways like small amphiphile additives successfully used in crystallization of integral membrane proteins, thereby decreasing the micellar radius and extruding water from the hydrophobic core of the micelles. The effect of these modifications on detergent micelle properties and on the stabilization and crystallization of integral membrane proteins is being investigated in collaboration with members of the Center for Innovative Membrane Protein Technologies of the Joint Center for Structural Genomics at Scripps Research. We are also interested in synthesizing additional novel amphiphilic molecules, including peptides, fluorinated lipids, and polymers that have special properties to facilitate the structural and functional study of integral membrane proteins. High-Throughput StructureBased Drug Discovery and Structural Neurobiology R.C. Stevens, E.E. Abola, A. Alexandrov, J.W. Arndt, G. Asmar-Rovira, R. Benoit, F. Bi, M.H. Bracey, D. Carlton, Q. Chai, J.C. Chappie, E. Chien, T. Clayton, B. Collins, A. Gámez, M. Griffith, C. Grittini, M.A. Hanson, A. Houle, J. Joseph, K. Masuda, B. McManus, K. Moy, M. Nelson, R. Page, M.G. Patch, C. Roth, K. Saikatendu, V. Sridhar, M. Straub, V. Subramanian, J. Velasquez, L. Wang, M. Yadav HIGH-THROUGHPUT STRUCTURAL BIOLOGY or the past several years, we have focused on developing tools to change the field of structural biology by accelerating the rate of determination of protein structures, an endeavor that includes pioneering microliter expression/purification for structural studies, nanovolume crystallization, and automated image collection. Applications of these technologies were initially tested at the Joint Center for Structural Genomics (http://www.jcsg.org), where we showed the power of the new tools. In addition to the recent funding of the JCSG-2 as a second-phase production center of the National Institute of General Medical Sciences, 2 new centers funded by the National Institutes of Health have been spun off for technologic innovations in structural biology. The first center is called the Joint Center for Innovative Membrane Protein Technologies (http://jcimpt.scripps.edu). Here, in collaboration with G. Chang, S. Lesley, K. Wüthrich, and Q. Zhang, Department of Molecular Biology; P. Kuhn F Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. and M. Yeager, Department of Cell Biology; and M.G. Finn, Department of Chemistry, we do research exclusively on membrane proteins, including G protein–coupled receptors. The second center is the Accelerated Technologies Center for Gene to 3D Structure (http://www .atcg3d.org). Here we are doing collaborative studies with P. Kuhn, Department of Cell Biology, and researchers from deCODE biostructures, Bainbridge Island, Washington; Lyncean Technologies, Palo Alto, California; and the University of Chicago, Chicago, Illinois. In the near future, this center will build a synchrotron resource at Scripps Research. STRUCTURAL NEUROBIOLOGY Although we have developed high-throughput methods to accelerate the determination of protein structures, our primary interest is using these tools to study the chemistry and biology of neurotransmission and of diseases that affect neurons. Our goals are to understand how neuronal cells function on a molecular level and, on the basis of that understanding, create new molecules and materials that mimic neuronal signal transduction and recognition. We use high-throughput protein crystallography and biochemical methods to probe the structure and function of molecules involved in neurotransmission and neurochemistry. FAT T Y A C I D A M I D E H Y D R O L A S E In collaboration with B.F. Cravatt, Department of Cell Biology, we solved the structure of fatty acid amide hydrolase (FAAH), a degradative integral membrane enzyme responsible for setting intracellular levels of endocannabinoids, to 2.8 Å. FAAH is intimately associated with CNS signaling processes such as retrograde synaptic transmission, a process that is also modulated by the illicit substance δ9-tetrahydrocannabinol. FAAH is a dimer capable of monotopic membrane insertion; it has an active-site structure consistent with the capacity for hydrolysis of hydrophobic fatty acid amides and structural features amenable to structure-based drug design. With our knowledge of the 3-dimensional structure, we are trying to understand how the enzyme works at a basic level and how it might be the basis for potential drug discovery. BIOSYNTHESIS OF NEUROTRANSMITTERS For neuronal signal transduction, the presynaptic cell synthesizes neurotransmitters that then traverse the synaptic cleft. We are using the high-throughput methods to determine the inclusive structures of complete biochemical pathways. Specifically, we are interested in determining the structures of all the enzymes MOLECULAR BIOLOGY 2005 in the biosynthesis pathways of neurotransmitters in order to understand the mechanistic details of each individual enzymatic reaction at the atomic level. This approach also allows us to determine the best path of drug discovery in the areas of neurotransmitter biosynthesis and catabolism. Phenylalanine hydroxylase and tyrosine hydroxylase initiate the first committed step in the biosynthesis of the neurotransmitters dopamine, adrenaline, and noradrenaline, and tryptophan hydroxylase catalyzes the rate-determining step in the biosynthesis of serotonin. Because of the importance of these neurotransmitters in the proper functioning of the CNS, understanding the molecular details involved in the catalysis and regulation of these biosynthetic enzymes is crucial. We determined the 3-dimensional structures for tyrosine hydroxylase, tryptophan hydroxylase, and phenylalanine hydroxylase, and we are uncovering specific mechanistic details for these enzymes. T H E R A P E U T I C A G E N T S F O R T R E AT M E N T O F PHENYLKETONURIA In addition to the basic hydroxylase enzymology questions under investigation, recent clinical studies suggest that some patients with the metabolic disease phenylketonuria are responsive to (6R)-L-erythro-5,6,7, 8-tetrahydrobiopterin, the natural cofactor of phenylalanine hydroxylase. We are doing studies to correlate how structure can be used to predict which patients with phenylketonuria most likely will respond to treatment with this cofactor. Currently, the proprietary form of the cofactor, Phenoptin, is entering phase 3 clinical trials for the treatment of mild phenylketonuria. For classical phenylketonuria, we are developing an enzyme replacement therapeutic agent that is being tested in animal models. The therapy is based on administration of a modified form of phenylalanine ammonia lyase discovered in our structural studies (Fig. 1). Last, we are determining the structural basis of diseases caused by several other enzymes involved in the biosynthesis of neurotransmitters. Many of these disorders are rare or occur during childhood. 175 F i g . 1 . A, Crystal structure of phenylalanine ammonia lyase (PAL) determined at 1.6-Å resolution. This protein structure was engineered and chemically modified as a potential once-a-week injectable therapeutic agent for treatment of phenylketonuria. B, ENU2 mice are used as a model for phenylketonuria in preclinical studies. C and D, Reduction in phenylalanine and immune response levels in ENU2 mice after the injection of PAL that has been chemically modified (pegylated). These PEG-PAL formulations show promise as therapeutic agents for treatment of phenylketonuria. NEUROTOXINS The clostridial neurotoxins include tetanus toxin and the 7 serotypes of botulinum toxin (Fig. 2). We are determining the molecular events involved in the binding, pore formation, translocation, and catalysis of botulinum neurotoxin. Although botulinum toxin is most known for its deadly effects, it is now being used therapeutically to treat involuntary muscle disorders. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . Serotype structures of botulinum neurotoxin (BoNT), its light chain (LC), and the closely related tetanus neurotoxin (TeNT). 176 MOLECULAR BIOLOGY 2005 Recently, we determined the structure of the 900-kD complex form of the toxin, the 150-kD holotoxin form, the catalytic domain, and the catalytic domain bound to substrates and inhibitors. These structures are being used to understand and redesign the toxin’s mechanism of action and to determine additional therapeutic applications of the toxin. PUBLICATIONS Arndt, J.W., Gu, J., Jaroszewski, L., Schwarzenbacher, R., Hanson, M.A., Lebeda, F.J., Stevens, R.C. The structure of the neurotoxin-associated protein HA33/A from Clostridium botulinum suggests a reoccurring β-trefoil fold in the progenitor toxin complex. J. Mol. Biol. 346:1083, 2005. Peti, W., Johnson, M.A., Hermann, T., Newman, B.W., Buchmeier, M.J., Nelson, M., Joseph, J., Page, R., Stevens, R.C., Kuhn, P., Wüthrich, K. Structural genomics of the severe acute respiratory syndrome coronavirus: nuclear magnetic resonance structure of the protein nsP7. J. Virol . 79:12905, 2005. Peti, W., Page, R., Wilson, I., Stevens, R., Wüthrich, K. Structural proteomics pipeline miniaturized using micro expression and microcoil NMR. J. Struct. Funct. Genomics, in press. Pey, A.L., Pérez, B., Desviat, L.R., Martinez, M.A., Aguado, C., Erlandsen, H., Gámez, A., Stevens, R.C., Thorolfsson, M., Ugarte, M., Martinez, A. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum. Mutat. 24:388, 2004. Ricci, J.S., Stevens, R.C., McMullan, R.K., Klooster, W.T. The crystal structure of strontium hydroxide octahydrate, Sr(OH)2.8H2O at 20, 100, and 200 K from neutron diffraction. Acta Crystrallogr. B 61:381. 2005. Arndt, J.W., Schwarzenbacher, R., Page, R., et al. Crystal structure of an α/β serine hydrolase (YDR428C) from Saccharomyces cerevisiae at 1.85 Å resolution. Proteins 58:755, 2005. Rife, C., Schwarzenbacher, R., McMullen, D., et al. Crystal structure of a putative modulator of DNA gyrase (pmbA) from Thermotoga maritima at 1.95 Å resolution reveals a new fold. Proteins 61:444, 2005. Arndt, J.W., Yu, W., Bi, F., Stevens, R.C. Crystal structure of botulinum neurotoxin type G light chain: serotype divergence in substrate recognition. Biochemistry 44:9574, 2005. Rife, C., Schwarzenbacher, R., McMullen, D., et al. Crystal structure of a global regulatory protein CsrA from Pseudomonas putida at 2.05 Å resolution reveals a new fold. Proteins 61:449, 2005. Cànaves, J.M., Page, R., Stevens, R.C. Protein biophysical properties that correlate with crystallization success in Thermotoga maritima: maximum clustering strategy for structural genomics. J. Mol. Biol. 344:977, 2004. Saikatendu, K.S., Joseph, J.S., Subramanian, V., Clayton, T., Griffith, M., Moy, K., Velasquez, J., Neuman, B.W., Buchmeier, M.J., Stevens, R.C., Kuhn, P. Structural basis of severe acute respiratory syndrome coronavirus (SARS-CoV) ADPribose-1′′-phosphate (Appr-1′′-p) dephosphorylation by a conserved domain of nsP3. Structure, in press. Carter, D.C., Rhodes, P., McRee, D.E., Tari, L.W., Dougan, D.R., Snell, G., Abola, E., Stevens, R.C. Reduction in diffuso-convective disturbances in nanovolume protein crystallization experiments. J. Appl. Crystrallogr. 38:87, 2005. Chappie, J.S., Cànaves, J.M., Han, G.W., Rife, C.L., Xu, Q., Stevens, R.C. The structure of a eukaryotic nicotinic acid phosphoribosyltransferase reveals structural heterogeneity among type II PRTases. Structure (Camb.) 13:1385, 2005. Erlandsen, H., Pey, A.L., Gámez, A., Pérez, B., Desviat, L.R., Aguado, C., Koch, R., Surendran, S., Tyring, T., Matalon, R., Scriver, C.R., Ugarte, M., Martínez, A., Stevens, R.C. Correction of kinetic and stability defects by the cofactor tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc. Natl. Acad. Sci. U. S. A. 101:16903, 2004. Gámez, A., Sarkissian, C.N., Wang, L., Kim, W., Straub, M., Patch, M.G., Chen, L., Striepeke, S., Fitzpatrick, P., Lemontt, J.F., O’Neill, C., Scriver, C.R., Stevens, R.C. Development of pegylated forms of recombinant Rhodosporidium toruloides phenylalanine ammonia-lyase for the treatment of classical phenylketonuria. Mol. Ther. 11:986, 2005. Han, G.W., Schwarzenbacher, R., Page, R., et al. Crystal structure of an alanineglyoxylate aminotransferase from Anabena sp at 1.70 Å resolution reveals a noncovalently linked PLP cofactor. Proteins 58:971, 2005. Levin, I., Miller, M.D., Schwarzenbacher, R., et al. Crystal structure of an indigoidine synthase A (IndA)-like protein (TM1464) from Thermotoga maritima at 1.90 Å resolution reveals a new fold. Proteins 59:864, 2005. Matalon, R., Michals-Matalon, K., Koch, R., Grady, J., Tyring, S., Stevens, R.C. Response of patients with phenylketonuria in the US to tetrahydrobiopterin. Mol. Genet. Metab., in press. Mathews, I., Schwarzenbacher, R., McMullan, D., et al. Crystal structure of S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) from Thermotoga maritima at 2.0 Å resolution reveals a new fold. Proteins 59:869, 2005. Page, R., Deacon, A.M., Lesley, S., Stevens, R.C. Shotgun crystallization strategy for structural genomics, II: crystallization and conditions that produce high resolution structures for T maritima proteins. J. Funct. Struct. Genomics 6:209, 2005. Scriver, C.R., Hurtubise, M., Prevost, L., Phommarinh, M., Konecki, D., Erlandsen, H., Stevens, R.C., Waters, P.J., Ryan, S., McDonald, D., Sarkissan C. A PAH gene knowledge base: content, informatics, utilization. In: PKU and BH4: Advances in Phenylketonuria and Tetrahydrobiopterin Research. Blaue, N. (Ed.), SPS Publications, Heilbrun, Germany, in press. Swaminathan, S., Stevens, R.C. Three-dimensional protein structures of botulinum neurotoxin light chains serotypes A, B, and E. In: Treatments from Toxins: The Therapeutic Potential of Clostridial Neurotoxins. Foster, K., Hambleton, P., Shone, C. (Eds.). CRC Press, Boca Raton, FL, in press. Wang, L., Gámez, A., Sarkissian, C.N., Straub, M., Patch, M.G., Han, G.W., Striepeke, S., Fitzpatrick, P., Scriver, C.R., Stevens, R.C. Structure-based chemical modification strategy for enzyme replacement treatment of phenylketonuria. Mol. Genet. Metab. 86:134, 2005. Xu, Q., Schwarzenbacher, R., McMullen, D., et al. Crystal structure of a formiminotetrahydrofolate cyclodeaminase (TM1560) from Thermotoga maritima at 2.80 Å resolution reveals a new fold. Proteins 58:976, 2005. Yadav, M.K., Gerdts, C.J., Sanishvili, R., Smith, W., Roach, L.S., Ismagilov, R.F., Kuhn, P., Stevens, R.C. In situ data collection and structure refinement from microcapillary protein crystallization. J. Appl. Crystallogr., in press. Nuclear Magnetic Resonance in Structural Biology and Structural Genomics K. Wüthrich, M. Almeida, L. Columbus, T. Etezady, M. Geralt, S. Hiller, R. Horst, M. Johnson, W.J. Placzek, Page, R., Peti, W., Wilson, I.A., Stevens, R.C., Wüthrich, K. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc. Natl. Acad. Sci. U. S. A. 102:1901, 2005. Pérez, B., Desviat, L.R., Gomez-Puertas, P., Martinez, A., Stevens, R.C., Ugarte, M. Kinetic and stability analysis of PKU mutations identified in BH4-responsive patients. Mol .Genet. Metab., in press. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. W. Peti, P. Serrano M embers of our laboratory participate in the Joint Center for Structural Genomics (JCSG), the JCSG Center for Innovative Membrane MOLECULAR BIOLOGY 2005 Protein Technologies, and the Functional and Structural Proteomics Analysis of SARS-CoV–Related Proteins Consortium. As part of these studies on protein structure, we develop and use nuclear magnetic resonance (NMR) methods to screen recombinant protein preparations for folded proteins. We are also exploring the use of microcoil NMR equipment combined with microexpression of proteins. We also use NMR spectroscopy to determine the structure of selected proteins from the proteomes under study in the structural genomics programs. Some of our research is described in the following sections. NMR SCREENING OF THERMOTOGA MARITIMA MEMBRANE PROTEINS A total of 45 predicted α-helical membrane proteins from Thermotoga maritima were selected as potential targets for solution NMR structural studies. These proteins have between 1 and 4 predicted helical transmembrane segments and have molecular weights less than 16 kD. Of the 45 targets, 10 were overexpressed in Escherichia coli, and 8 of these 10 localized to the bacterial membrane. These 8 protein targets were purified and screened to determine efficient detergents for solubilization. To evaluate the fold and the aggregation state of the proteins in the best conditions thus identified, we used 1-dimensional 1H NMR spectroscopy to screen the targets. For 3 of the 8 proteins, the NMR spectra indicated soluble protein-detergent complexes. The transverse relaxation optimized spectroscopy correlation spectra of these 3 targets provided evidence that these 3 proteins are folded helical proteins. Experiments are under way for NMR assignment and structure determination of these α-helical membrane proteins in mixed micelles with detergents. S T R U C T U R E D E T E R M I N AT I O N S O F C O N S E R V E D HYPOTHETICAL PROTEINS FROM T MARITIMA The NMR structure of the conserved hypothetical protein TM1816 from T maritima has an α/β topology with 3 α-helices and a 5-stranded β-sheet. The molecular architecture of TM1816 is similar to that of 2 other conserved hypothetical proteins, TM1290 from T maritima (33% sequence identity) and MTH1175 from Methanobacterium thermoautotrophicum (30% sequence identity). These 3 proteins belong to the cluster of orthologous groups 1433 and are structurally similar to the Azobacter vinelandii iron, molybdenum cofactor-binding protein NafY. TM1816 is unique among the 3 homologs because it contains a histidine residue Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 177 corresponding to the one that is crucial for cofactor binding in NafY. TM0487 is a 104-residue protein from T maritima that was identified via NMR screening as a potential target for NMR structure determination. The 3-dimensional structure of TM0487 provides a foundation for functional studies of an entire class of proteins, because TM0487 has a large number of homologs on the amino acid sequence level, including 216 nonredundant sequences that contain a type 59 domain of unknown function. So far, a 3-dimensional structure has not been determined for any of these homologous proteins. The conserved residues among the aforementioned 216 sequences are clustered in the hydrophobic core of the TM0487 fold and in a putative active site exposed to the solvent. Overall, strong evidence indicates that the TM0487 fold is preserved in all of this class of domains of unknown function, so that this structure determination provides a foundation for focused functional studies of a wide variety of otherwise so far only minimally characterized proteins. NMR STUDIES OF AN ACYL CARRIER PROTEIN F R O M T H E C YA N O B A C T E R I U M A N A B A E N A Asl1650, a protein obtained from the cyanobacterium Anabaena, was identified as an ortholog of a mouse protein domain as part of a JCSG bioinformatics strategy to extend information on the protein folding space of eukaryotic proteins. The protein was selected for NMR structure determination on the basis of an NMR screen of recombinant mouse protein homologs expressed in E coli. Acyl carrier proteins (ACPs) are central components of complex multienzyme systems that function in the metabolism of all living organisms. These systems catalyze the biosynthesis of fatty acids, signaling molecules, and bioactive natural products. The polyketide synthases and nonribosomal peptide synthetases of microorganisms produce compounds with antibiotic and anticancer activities. An understanding of structure-function relationships in these widely distributed enzyme systems is thus of obvious interest for the design of new therapeutic compounds. The protein Asl1650 is only distantly related to previously characterized ACPs. It was derived from Anabaena sp PCC 7120, a filamentous cyanobacterium. Members of this genus of cyanobacteria produce a variety of bioactive compounds, which are as yet only poorly characterized. We determined the solution structure of Asl1650 by using high-resolution NMR 178 MOLECULAR BIOLOGY 2005 spectroscopy. The structure had a surprising similarity to the structures of peptidyl carrier protein domains, which usually occur as single domains of giant, multifunctional proteins. A variant active-site sequence, asparagine–serine–serine, occurs in similar orientation to the aspartic acid–serine–leucine sequence of known ACPs. These structural similarities suggest that Asl1650 may function as a discrete peptidyl carrier protein domain in a nonribosomal peptide synthetase pathway or a hybrid polyketide synthase–nonribosomal peptide synthetase pathway. PUBLICATIONS Almeida, M.S., Peti, W., Wüthrich, K. 1H-, 13C- and 15N-NMR assignment of the conserved hypothetical protein TM0487 from Thermotoga maritima. J. Biomol. NMR 29:453, 2004. Etezady-Esfarjani, T., Herrmann, T., Peti, W., Klock, H.E., Lesley, S.A., Wüthrich, K. NMR structure determination of the hypothetical protein TM1290 from Thermotoga maritima using automated NOESY analysis. J. Biomol. NMR 29:403, 2004. Page, R., Peti, W., Wilson, I.A., Stevens, R.C., Wüthrich, K. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc. Natl. Acad. Sci. U. S. A. 102:1901, 2005. Peti, W., Etezady-Esfarjani, T., Herrmann, T., Klock, H.E., Lesley, S.A., Wüthrich, K. NMR for structural proteomics of Thermotoga maritima: screening and structure determination. J. Struct. Funct. Genomics 5:205, 2004. Peti, W., Norcross, J., Eldridge, G., O’Neil-Johnson, M. Biomolecular NMR using a microcoil NMR probe: new technique for the chemical shift assignment of aromatic side chains in proteins. J. Am. Chem. Soc. 126:5873, 2004. Nuclear Magnetic Resonance of 3-Dimensional Structure and Dynamics of Proteins in Solution P.E. Wright, H.J. Dyson, R. Burge, R. De Guzman, T. Dunzendorfer-Matt, J. Ferreon, N. Greenman, T.-H. Huang, M. Kostic, J. Lansing, B. Lee, M. Landes, M. Martinez-Yamout, T. Nishikawa, J. Wojciak, M. Zeeb, E. Manlapaz, L.L. Tennant, J. Chung, D.A. Case, J. Gottesfeld, R. Evans,* M. Montminy* * Salk Institute, La Jolla, California e use multidimensional nuclear magnetic resonance (NMR) spectroscopy to investigate the structures, dynamics, and interactions of proteins in solution. Such studies are essential for understanding the mechanisms of action of these proteins and for elucidating structure-function relationships. The focus of our current research is protein-protein and protein–nucleic acid interactions involved in the regulation of gene expression. W Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. TRANSCRIPTION FACTOR–NUCLEIC ACID COMPLEXES NMR methods are being used to determine the 3-dimensional structures and intramolecular dynamics of zinc finger motifs from several eukaryotic transcriptional regulatory proteins, both free and complexed with target nucleic acid. Zinc fingers are among the most abundant domains in eukaryotic genomes. They play a central role in the regulation of gene expression at both the transcriptional and the posttranscriptional levels, mediated through their interactions with DNA, RNA, or protein components of the transcriptional machinery. The C2H 2 zinc finger, first identified in transcription factor IIIA (TFIIIA), is used by numerous transcription factors to achieve sequence-specific recognition of DNA. There is growing evidence, however, that some C 2H 2 zinc finger proteins control gene expression both through their interactions with DNA regulatory elements and, at the posttranscriptional level, by binding to RNA. The best-characterized example of a C2H 2 zinc finger protein that binds specifically to both DNA and to RNA is TFIIIA, which contains 9 zinc fingers. We showed previously that different subsets of zinc fingers are responsible for high-affinity binding of TFIIIA to DNA (fingers 1–3) and to 5S RNA (fingers 4–6). To obtain insights into the mechanism by which the TFIIIA zinc fingers recognize both DNA and RNA, we are using NMR methods to determine the structures of the complex formed by zf1-3 (a protein containing fingers 1–3) with DNA and by zf4-6 (a protein consisting of fingers 4–6) with a fragment of 5S RNA. Three-dimensional structures were determined previously for the complex of zf1-3 with the cognate 15-bp oligonucleotide duplex. The structures contain several novel features and reveal that prevailing models of DNA recognition, which assume that zinc fingers are independent modules that contact bases through a limited set of amino acids, are outmoded. In addition to its role in binding to and regulating the 5S RNA gene, TFIIIA also forms a complex with the 5S RNA transcript. We recently determined the NMR structure of the complex formed by zinc fingers 4–6 with a truncated form of 5S RNA (Fig. 1). The structure has provided important insights into the structural basis for 5S RNA recognition. Finger 4 of the protein recognizes both the structure of the RNA backbone and the specific bases in the loop E motif of the RNA, in a classic lockand-key interaction. Fingers 5 and 6, with a single residue between them, undergo mutual induced-fit folding with the loop A region of the RNA, which is highly flexible in the absence of the protein. MOLECULAR BIOLOGY 2005 179 ment. This structure showed sequence-specific recognition of single-stranded RNA through formation of a network of hydrogen bonds between the polypeptide backbone and the Watson-Crick edges of the bases. PROTEIN-PROTEIN INTERACTIONS IN T R A N S C R I P T I O N A L R E G U L AT I O N F i g . 1 . Structure of zinc fingers 4–6 of TFIIIA bound to 5S RNA. The protein backbone is shown as a ribbon, and the phosphate backbone and bases of the RNA are displayed as gray tubes. NMR studies of 2 alternate splice variants of the Wilms tumor zinc finger protein are in progress. These proteins differ only through insertion of 3 additional amino acids (the tripeptide lysine-threonine-serine) in the linker between fingers 3 and 4, yet have marked differences in their DNA-binding properties and subcellular localization. 15 N relaxation measurements indicate that the insertion increases the flexibility of the linker between fingers 3 and 4 and abrogates binding of the fourth zinc finger to its cognate site in the DNA major groove, thereby modulating DNA-binding activity. The x-ray structure of the DNA complex has now been determined, and NMR studies of RNA binding are in progress. We have also determined the structure of the first member of a novel class of C2H 2 zinc finger proteins that bind specifically to double-stranded RNA. Several novel zinc binding motifs have recently been identified that mediate gene expression at the posttranscriptional level by regulating mRNA processing and metabolism. Regulatory proteins of the TIS11 family bind specifically, through a pair of novel CCCH zinc fingers, to the adenosine-uridine–rich element in the 3′ untranslated region of short-lived cytokine, growth factor, and protooncogene mRNAs and control expression by promoting rapid degradation of the message. We recently determined the NMR structure of the complex formed between the tandem zinc finger domain of TIS11d and its binding site on the adenosine-uridine–rich elePublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Transcriptional regulation in eukaryotes relies on protein-protein interactions between DNA-bound factors and coactivators that, in turn, interact with the basal transcription machinery. The transcriptional coactivator CREB-binding protein (CBP) and its homolog p300 play an essential role in cell growth, differentiation, and development. Understanding the molecular mechanisms by which CBP and p300 recognize their various target proteins is of fundamental biomedical importance. CBP and p300 have been implicated in diseases such as leukemia, cancer, and mental retardation and are novel targets for therapeutic intervention. We previously determined the structure of the kinaseinducible activation domain of the transcription factor CREB bound to its target domain (the KIX domain) in CBP. Ongoing work is directed toward mapping the interactions between KIX and the transcriptional activation domains of the proto-oncogene c-Myb and of the mixedlineage leukemia protein. The solution structure of the ternary complex composed of KIX, c-Myb, and the mixedlineage leukemia protein has been completed (Fig. 2) and provides insights into the structural basis for the ability of the KIX domain to interact simultaneously and allosterically with 2 different effectors. Our work has also provided new understanding of the thermodynamics of the coupled folding and binding processes involved in interaction of KIX with transcriptional activation domains. F i g . 2 . Structure of the ternary complex between the KIX domain of CBP (pale gray) and the transcriptional activation domains of c-Myb and the mixed-lineage leukemia protein (MLL). 180 MOLECULAR BIOLOGY 2005 Recently, we determined the structure of the complex between the hypoxia-inducible factor Hif-1α and the CH1 domain of CBP. The interaction between Hif-1α and CBP/p300 is of major therapeutic interest because of the central role Hif-1α plays in tumor progression and metastasis; disruption of this interaction leads to attenuation of tumor growth. A protein named CITED2 functions as a negative feedback regulator of the hypoxic response by competing with Hif-1α for binding to the CH1 domain of CBP. We determined the structure of the complex formed between CITED2 and the CH1 domain and were able to show that the CH1 domain is folded into a stable 3-dimensional structure even in the absence of binding partners. The intrinsically unstructured Hif-1α and CITED2 domains use partly overlapping surfaces of the CH1 motif to achieve high-affinity binding and compete effectively with each other for CBP/p300. The structure of another zinc-binding module of CBP, the ZZ domain, has a novel fold (Fig. 3), but its function is not yet understood. We are continuing to map the multiplicity of interactions between CBP/p300 domains and their numerous biological targets to understand the complex interplay of interactions that mediate key biological processes in health and disease. Legge, G.B., Martinez-Yamout, M.A., Hambly, D.M., Trinh, T., Lee, B.M., Dyson, H.J., Wright, P.E. ZZ domain of CBP: an unusual zinc finger fold in a protein interaction module. J. Mol. Biol. 343:1081, 2004. Möller, H.M., Martinez-Yamout, M.A., Dyson, H.J. Wright, P.E. Solution structure of the N-terminal zinc fingers of the Xenopus laevis double-stranded RNA-binding protein ZFa. J. Mol. Biol. 351:718, 2005. Folding of Proteins and Protein Fragments P.E. Wright, H.J. Dyson, C. Nishimura, D. Felitsky, Y. Yao, J. Chung, L.L. Tennant, V. Bychkova* * Institute of Protein Research, Puschino, Russia he molecular mechanism by which proteins fold into their 3-dimensional structures remains one of the most important unsolved problems in structural biology. Nuclear magnetic resonance (NMR) spectroscopy is uniquely suited to provide information on the structure of transient intermediates formed during protein folding. Previously, we used NMR methods to show that many peptide fragments of proteins have a tendency to adopt folded conformations in water solution. The presence of transiently populated folded structures, including reverse turns, helices, nascent helices, and hydrophobic clusters, in water solutions of short peptides has important implications for initiation of protein folding. Formation of elements of secondary structure probably plays an important role in the initiation of protein folding by reducing the number of conformations that must be explored by the polypeptide chain and by directing subsequent folding pathways. T A P O M Y O G L O B I N F O L D I N G PAT H WAY F i g . 3 . Structure of the ZZ zinc finger domain of CBP. PUBLICATIONS De Guzman, R.N., Goto, N.K., Dyson, H.J., Wright, P.E. Structural basis for cooperative transcription factor binding to the CBP coactivator. J. Mol. Biol., in press. De Guzman, R.N., Wojciak, J.M., Martinez-Yamout, M.A., Dyson, H.J., Wright, P.E. CBP/p300 TAZ1 domain forms a structural scaffold for ligand binding. Biochemistry 44:490, 2005. Dyson, H.J., Wright, P.E. Intrinsically unstructured proteins and their function. Nat. Rev. Mol. Cell Biol. 6:197, 2005. Gearhart, M.D., Dickinson, L., Ehley, J., Melander, C., Dervan, P.B., Wright, P.E., Gottesfeld, J.M. Inhibition of DNA binding by human estrogen related receptor-2 and estrogen receptor α with minor groove binding polyamides. Biochemistry 44:4196, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. A major program in our laboratory is directed toward a structural and mechanistic description of the apomyoglobin folding pathway. Previously, we used quenched-flow pulse labeling methods in conjunction with 2-dimensional NMR spectroscopy to map the kinetic folding pathway of the wild-type protein. With these methods, we showed that an intermediate in which the A, G, and H helices adopt hydrogen-bonded secondary structure is formed within 6 ms of the initiation of refolding. Folding then proceeds by stabilization of structure in the B helix and then in the C and E helices. We are using carefully selected myoglobin mutants and both optical stopped-flow spectroscopy and NMR methods to further probe the kinetic folding pathway. For some of the mutants studied, the changes in amino acid sequence resulted in changes in the fold- MOLECULAR BIOLOGY 2005 ing pathway of the protein. These experiments are providing novel insights into both the local and the longrange interactions that stabilize the kinetic folding intermediate. Of particular importance, long-range interactions have been observed that indicate nativelike packing of some of the helices in the kinetic molten globule intermediate. Apomyoglobin provides a unique opportunity for detailed characterization of the structure and dynamics of a protein-folding intermediate. Conditions were previously identified under which the apomyoglobin molten globule intermediate is sufficiently stable for acquisition of multidimensional heteronuclear NMR spectra. Analysis of 13C and other chemical shifts and measurements of polypeptide dynamics provided unprecedented insights into the structure of this state. The A, G, and H helices and part of the B helix are folded and form the core of the molten globule. This core is stabilized by relatively nonspecific hydrophobic interactions that restrict the motions of the polypeptide chain. Fluctuating helical structure is formed in regions outside the core, although the population of helix is low and the chain retains considerable flexibility. The F helix acts as a gate for heme binding and only adopts stable structure in the fully folded holoprotein. The acid-denatured (unfolded) state of apomyoglobin is an excellent model for the fluctuating local interactions that lead to the transient formation of unstable elements of secondary structure and local hydrophobic clusters during the earliest stages of folding. NMR data indicated substantial formation of helical secondary structure in the acid-denatured state in regions that form the A and H helices in the folded protein and also revealed nonnative structure in the D and E helix region. Because the A and H regions adopt stabilized helical structure in the earliest detectable folding intermediate, these results lend strong support to folding models in which spontaneous formation of local elements of secondary structure plays a role in initiating formation of the A-[B]-G-H molten globule folding intermediate. In addition to formation of transient helical structure, formation of local hydrophobic clusters has been detected by using 15N relaxation measurements. Significantly, these clusters are formed in regions where the average surface area buried upon folding is large. In contrast to acid-denatured unfolded apomyoglobin, the ureadenatured state is largely devoid of structure, although residual hydrophobic interactions have been detected by using relaxation measurements. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 181 We measured residual dipolar couplings for unfolded states of apomyoglobin by using partial alignment in strained polyacrylamide gels. These data provide novel insights into the structure and dynamics of the unfolded polypeptide chain. We have shown that the residual dipolar couplings arise from the well-known statistical properties of flexible polypeptide chains. Residual dipolar couplings provide valuable insights into the dynamic and conformational propensities of unfolded and partly folded states of proteins and hold great promise for charting the upper reaches of protein-folding landscapes. To probe long-range interactions in unfolded and partially folded states of apomyoglobin, we introduced spin-label probes at several sites throughout the polypeptide chain. These experiments led to the surprising discovery that structures with nativelike topology exist within the ensemble of conformations formed by the acid-denatured state of apomyoglobin. They also indicated that the packing of helices in the molten globule state is similar to that in the native folded protein. The view of protein folding that results from our work on apomyoglobin is one in which collapse of the polypeptide chain to form increasingly compact states leads to progressive accumulation of secondary structure and increasing restriction of fluctuations in the polypeptide backbone. Chain flexibility is greatest at the earliest stages of folding, in which transient elements of secondary structure and local hydrophobic clusters are formed. As the folding protein becomes increasingly compact, backbone motions become more restricted, the hydrophobic core is formed and extended, and nascent elements of secondary structure are progressively stabilized. The ordered tertiary structure characteristic of the native protein, with well-packed side chains and relatively low-amplitude local dynamics, appears to form rather late in folding. We recently introduced a variation on the classic quench-flow technique, which makes use of the capabilities of modern NMR spectrometers and heteronuclear NMR experiments, to study the proteins labeled along the folding pathway in an unfolded state in an aprotic organic solvent. This method allows detection of many more amide proton probes than in the classic method, which required formation of the fully folded protein and the measurement of the protein’s NMR spectrum in water solutions (Fig. 1). This method is particularly useful in documenting changes in the folding pathway that result in the destabilization of parts of the protein in the molten globule intermediate. We recently 182 MOLECULAR BIOLOGY 2005 F i g . 1 . High-resolution view of the backbone structure of the 6.4-ms burst-phase kinetic folding intermediate of apomyoglobin. The tube thickness and darkness indicate the extent of folding into helical structure. Helices that are fully folded are indicated by thick, dark tubes. Regions that are partly folded are intermediate in thickness and shade, and regions of the protein that remain fully unstructured in the kinetic intermediate are represented by thin lines. introduced self-compensating mutations designed to change the amino acid sequence such that the average area buried upon folding in the A and E helix regions is significantly changed, while the 3-dimensional structure of the final folded state remains the same. These studies indicated that the average area buried upon folding is an accurate predictor of those parts of the apomyoglobin molecule that will fold first and participate in the molten globule intermediate (Fig. 2). FOLDING-UNFOLDING TRANSITIONS IN CELLULAR M E TA B O L I S M Many species of bacteria sense and respond to their own population density by an intricate autoregulatory mechanism known as quorum sensing; the bacteria release extracellular signal molecules, called autoinducers, for cell-cell communication within and between bacterial species. A number of bacteria appear to use quorum sensing for regulation of gene expression in response to fluctuations in cell population density. Processes regulated in this way include symbiosis, virulence, competence, conjugation, production of antibiotics, motility, sporulation, and formation of biofilms. We determined the 3-dimensional solution structure of a complex composed of the N-terminal 171 residues of the quorum-sensing protein SdiA of Escherichia coli and an autoinducer molecule, N-octanoyl-1-homoserine lactone (HSL). The SdiA-HSL system shows the “folding switch” behavior associated with quorum-sensing factors produced by other bacterial species. In the presence of HSL, the SdiA protein is stable and folded Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . Correlation between average surface area buried upon folding (AABUF, gray line) and regions of apomyoglobin that are folded in the kinetic burst-phase intermediate. Folded regions are indicated by high values of the proton occupancy (A0, black circles). Data are shown for the wild-type protein (A) and for a mutant protein (B) in which hydrophobic residues are moved from the A helix into the E helix region, thereby changing the folding pathway in a predictable manner. and can be produced in good yields from an E coli expression system. In the absence of the autoinducer, the protein is expressed into inclusion bodies. Samples of the SdiA-HSL complex can be denatured but cannot be refolded in aqueous buffers. The solution structure of the complex provides a likely explanation for this behavior. The autoinducer molecule is tightly bound in a deep pocket in the hydrophobic core and is bounded by specific hydrogen bonds to the side chains of conserved residues. The autoinducer thus forms an integral part of the hydrophobic core of the folded SdiA. PUBLICATIONS Dyson, H.J., Wright, P.E. Elucidation of the protein folding landscape by NMR. Methods Enzymol. 394:299, 2005. Dyson, H.J., Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6:197, 2005. Nishimura, C., Dyson, H.J., Wright, P.E. Enhanced picture of protein-folding intermediates using organic solvents in H/D exchange and quench-flow experiments. Proc. Natl Acad. Sci. U. S. A. 102:4765, 2005. Nishimura, C., Dyson, H.J. Wright, P.E. Identification of native and nonnative structure in kinetic folding intermediates of apomyoglobin. J. Mol. Biol., in press. Nishimura, C., Lietzow, M.A., Dyson H.J., Wright, P.E. Sequence determinants of a protein folding pathway. J. Mol. Biol. 351:383, 2005. MOLECULAR BIOLOGY 2005 Nuclear Magnetic Resonance Studies of the Structure and Dynamics of Enzymes H.J. Dyson, P.E. Wright, D. Boehr, M.O. Ebert, G. Kroon, J. Lansing, C.W. Lee, M. Martinez-Yamout, D. McElheny, N.E. Preece, K. Sugase, H.S. Won, Y. Yao, L.L. Tennant, J. Chung, C.L. Brooks, S.J. Benkovic,* A. Holmgren** * Pennsylvania State University, University Park, Pennsylvania ** Karolinska Institutet, Stockholm, Sweden e use site-specific information from nuclear magnetic resonance (NMR) to further the understanding of enzyme function through study of enzyme structure and dynamics. We focus on the mechanisms of enzymes and the relationship between dynamics and function in cellular control by thiol-disulfide chemistry. W DYNAMICS IN ENZYME ACTION Dynamic processes are implicit in the catalytic function of all enzymes. We use state-of-the-art NMR methods to elucidate the dynamic properties of several enzymes. New methods have been developed for analysis of NMR relaxation data for proteins that tumble anisotropically and for analysis of slow time scale motions. Dihydrofolate reductase plays a central role in folate metabolism and is the target enzyme for a number of anticancer agents. 15N relaxation experiments on dihydrofolate reductase from Escherichia coli revealed a rich diversity of backbone dynamics for a broad range of time scales (picoseconds to milliseconds). These studies were extended to additional intermediates in the reaction cycle and to forms of the enzyme with mutations at various motional “hot spots.” In addition, we are using 2H relaxation measurements in triple-labeled dihydrofolate reductase to elucidate the dynamics of critical active-site side chains. So far, we have identified functionally important motions in loops that control access to the active site of the reductase on the same time scale as the hydride transfer chemistry. These motions become attenuated once the NADPH cofactor is bound in the active site, locking the nicotinamide ring in a geometry conducive to hydride transfer to substrate. We also found evidence of motion of active-site side chains that are implicated in the catalytic process. Most recently, we used relaxation dispersion measurements to obtain direct information on microsecondPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 183 millisecond time scale motions in dihydrofolate reductase, allowing us to characterize the structures of the excited states involved in some of these catalysis-relevant processes. Fluctuations between these states, which involve motions of the nicotinamide ring of the cofactor into and out of the active site, occur on a time scale that is directly relevant to the structural transitions involved in progression through the catalytic cycle. Dihydrofolate reductase is also the test system for a series of experiments to address the question, If all of the chemistry goes on at the active site, what is the purpose of the rest of the enzyme? We will use a series of chimeric mutants, synthesized by our collaborator S.J. Benkovic, Pennsylvania State University, by using a library approach. The purpose of these experiments is to test the hypothesis that local variations in amino acid sequence, 3-dimensional structure, and polypeptide chain dynamics strongly influence the local interactions that mediate enzyme catalysis and may constitute the essential circumstance that allows enzymes to achieve high turnover rates as well as exquisite specificity in their reactions. A combination of NMR structure and dynamics measurements, single-molecule fluorescence measurements, and analysis of the catalytic steps in these mutant proteins will provide new insights into the role of the protein in enzyme catalysis. REDOX CONTROL BY THIOL-DISULFIDE CHEMISTRY Many cellular functions are regulated by thiol-disulfide chemistry. The importance of redox chemistry, particularly disulfide-dithiol equilibria, in cellular control mechanisms has only recently been recognized. For example, the chaperone heat-shock protein 33 (Hsp33) is regulated by a redox switch; the C-terminal domain of Hsp33 contains cysteines that are reduced and bound to zinc under normoxic conditions, but upon oxidation, the zinc is lost and disulfide bonds form. Interestingly, the zinc-bound form of the C-terminal domain is well structured, with a distinctive fold. NMR studies revealed that upon oxidation, the C-terminal domain becomes unstructured. We think that this loss of local structure exposes a dimerization site. Thus, under oxidative stress conditions, the chaperone dimerizes to the active form. We did an extensive study of the structural basis for the activity of several thiol-disulfide enzymes. Thioredoxin, a small, 108-residue thiol-disulfide oxidoreductase, has many functions in the cell, including reduction of ribonucleotides to form deoxyribonucleotides for DNA synthesis. A primary function of thioredoxin in the cell is as a protein disulfide reductase, a function vital for 184 MOLECULAR BIOLOGY 2005 the prevention of misfolded proteins in vivo. The E coli thioredoxin system has been fully characterized by using NMR, including the calculation of high-resolution structures for both the oxidized (disulfide) and the reduced (dithiol) forms of the protein. Using backbone dynamics and amide proton hydrogen exchange, we found that functional differences in phage systems between oxidized and reduced thioredoxin were due to differences in the flexibility of the molecules rather than to structural differences. We also delineated the mechanism of E coli thioredoxin. We found that the reduction reaction of thioredoxin depends critically on the movement of protons, during the 2-electron–2-proton transfer reaction, as a substrate disulfide is reduced. We are investigating a variant E coli thioredoxin with an N-terminal extension that binds zinc. This exciting new molecule may be another example of a redox-active, zinc-binding protein, previously exemplified by the redoxswitch domain of the chaperone Hsp33. Glutaredoxins are another major class of thiol-disulfide regulatory proteins. We recently determined the structure of glutaredoxin-2 from E coli. This protein appears to be a link between the glutaredoxin-thioredoxin class of small thiol-active proteins and the extensive glutathione-S-transferase class of detoxification enzymes. Glutaredoxins are thought to be involved in the processes that result in the attachment and removal of glutathione and nitrosyl groups from redox-active proteins. These processes, together with the formation of disulfide bonds, regulate the activity of redox-active proteins such as the transcription factor OxyR, which we also study. PUBLICATIONS Chen, J., Won, H.-S., Im, W., Dyson, H.J., Brooks, C.L. III. Generation of nativelike protein structures from limited NMR data, modern force fields and advanced conformational sampling. J. Biomol. NMR 31:59, 2005. McElheny, D., Schnell, J.R., Lansing, J.C., Dyson, H.J., Wright, P.E. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. U. S. A. 102:5032, 2005. Venkitakrishnan, R.P., Zaborowski, E., McElheny, D., Benkovic, S.J., Dyson, H.J., Wright, P.E. Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry 43:16046, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Ring Assemblies Mediating ATP-Dependent Protein Folding and Unfolding A.L. Horwich, W.A. Fenton, E. Chapman, E. Koculi arge ring assemblies function in many cellular contexts as compartments within a compartment, where actions can be carried out on a substrate bound in the central space inside an oligomeric ring by a high local concentration of surrounding active sites. Both protein folding and unfolding are carried out in an ATP-dependent fashion by such assemblies. We are studying the essential double-ring components, chaperonins, that assist protein folding to the native state. We are focusing on the bacterial chaperonin GroEL and more recently have been examining an opposite number, an “unfoldase,” the bacterial heat-shock protein 100 ring assembly known as ClpA. In the past year, we focused on polypeptide binding and ATPmediated action by both machines, showing quite different mechanisms. L G R O E L - M E D I AT E D F O L D I N G We are investigating polypeptide binding by an open ring of GroEL that is mediated through contacts between the exposed hydrophobic surface of nonnative polypeptide and a hydrophobic lining of the open ring. This step is one that potentially mediates unfolding of kinetically trapped states. In collaboration with K. Wüthrich, Department of Molecular Biology, using solution nuclear magnetic resonance and transverse relaxation optimized spectroscopy, we examined the structure of isotopelabeled human dihydrofolate reductase bound to GroEL. The resonances detected indicate that the reductase does not occupy a stable tertiary structure while bound to an open GroEL ring and also suggest that the enzyme is undergoing conformational exchange. This unfolded state was, however, productive; upon addition of ATP and the cochaperonin GroES, a nativelike pattern of resonances was recovered. The binding of ATP and GroES triggers productive GroEL-GroES–mediated folding in the encapsulated now-hydrophilic cavity of the GroES-bound ring (Fig. 1). By contrast, addition of ADP and GroES does not trigger folding. Surprisingly, however, x-ray and solution electron cryomicroscopy structures of GroEL-GroESADP and GroEL–GroES–ADP–aluminum fluoride, which is a folding-active state, are isomorphous. We noted MOLECULAR BIOLOGY 2005 185 with GroEL-polypeptide complexes in ADP, evidently forming a collision complex, but subsequent apical movement is impaired. We are using electron microscopy to examine the putative collision state, because it most likely is a state that is transiently populated in the physiologic nucleotide ATP. C L PA - M E D I AT E D U N F O L D I N G F i g . 1 . Protein folding and unfolding by chaperone ring assemblies. In protein folding mediated by the chaperonin GroEL (left), the energy of binding ATP and the cochaperonin GroES are used to produce rigid body movements of a GroEL ring that eject a bound nonnative substrate polypeptide into a GroES-encapsulated central cavity, switched from hydrophobic (shaded) to hydrophilic wall character, where productive folding proceeds. The free energy provided by a set of hydrogen bonds formed between the γ-phosphate of ATP and the nucleotide pocket is critical to producing a power stroke of apical domain movement that can eject the substrate polypeptide into the folding chamber. In contrast, in ClpA-mediated unfolding (right), this chaperone seems to use ATP hydrolysis by its D2 ATPase domain to drive a forceful distalward movement of a loop facing its central channel, exerting mechanical force on a bound protein that is proposed to exert an unfolding action. that these structure determinations were all carried out in the absence of substrate polypeptide and that a bound substrate potentially represents a load on the ring to which it is bound, resisting nucleotide/GroESdriven elevation and twist of the apical domain that are associated with ejection of a bound polypeptide off the cavity wall into the GroES-encapsulated cavity where productive folding occurs. Thus, the γ-phosphate of ATP might be critical to exerting a power stroke of apical movement. Consistent with such an idea, we found that addition of aluminum fluoride to a GroEL-GroES-ADPpolypeptide complex triggered productive folding. Further, we found that a substantial amount of free energy was released upon binding of aluminum fluoride to GroEL-GroES-ADP. To directly monitor apical movement, we used fluorescence resonance energy transfer between a fluorophore placed on the stable equatorial base of a subunit and a fluorophore placed in the apical domain (at a position that moves ~30 Å during the transition of a ring from unbound to GroES bound). Indeed, when no substrate was present, the apical domains opened rapidly (<1 second) upon addition of either ADP-GroES or ATP-GroES. In contrast, in the presence of bound polypeptide, only ATP-GroES could promote such rapid opening; ADP-GroES was either unable to drive opening at all or required a longer time (>40 seconds). Additional studies with fluorophores placed on GroEL and GroES indicated that GroES can associate rapidly Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. ClpA recognizes terminal peptide tags of proteins that are concordantly unfolded and translocated through its central channel. The polypeptide is generally directly translocated into a double-ring proteasome-like protease, ClpP, where it is degraded. During this past year, we used chemical cross-linkers placed on tag elements to identify channel-facing structures of ClpA that bind the tags and then did mutational analysis of the identified regions. For example, the C-terminal 11-residue ssrA peptide, which is added to proteins stalled at the ribosome to recruit these chains to ClpA, binds to 3 loops in the central channel of ClpA, 2 at the level of the proximal D1 ATPase domain and 1 at the level of the distal D2 ATPase (Fig. 1). Interestingly, a mutation at the point of insertion of the D2 loop into the channel wall allows substrate binding but blocks unfolding/ translocation, suggesting that this loop, connected to the more active D2 ATPase of ClpA, is a translocator that pulls on bound polypeptide in association with ATP hydrolysis, exerting a mechanical force that mediates unfolding. Consistently, x-ray studies of different nucleotide states have shown that 2 other such ring components that act on nucleic acids, the phi12 packaging motor and simian virus 40 T antigen, undergo such movements of channel-facing loops. PUBLICATIONS Hinnerwisch, J., Fenton, W.A., Furtak, K., Farr, G.W., Horwich, A.L. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell 121:1029, 2005. Horst, R., Bertelsen, E.B., Fiaux, J., Wider, G., Horwich, A.L., Wüthrich, K. Direct NMR observation of a substrate protein bound to the chaperonin GroEL. Proc. Natl. Acad. Sci. U. S. A. 102:000, 2005. Motojima, F., Chaudhry, C., Fenton, W.A., Farr, G.W., Horwich, A.L. Substrate polypeptide presents a load on the apical domains of the chaperonin GroEL. Proc. Natl. Acad. Sci. U. S. A. 101:15005, 2004. 186 MOLECULAR BIOLOGY 2005 Chemical Regulation of Gene Expression J.M. Gottesfeld, D. Alvarez-Carbonell, R. Burnett, J. Chou, D. Herman, K. Jennsen, S. Ku, P.B. Dervan*, K. Luger** * California Institute of Technology, Pasadena, California ** Colorado State University, Fort Collins, Colorado T R A N S C R I P T I O N R E G U L AT I O N W I T H S M A L L MOLECULES yrrole-imidazole polyamides are the only available class of synthetic small molecules that can be designed to bind predetermined DNA sequences with affinities comparable to those of cellular gene regulatory proteins. In collaboration with P.B. Dervan and colleagues at the California Institute of Technology, we showed that polyamides inhibit the DNA-binding activities of various transcriptional regulatory proteins and can be used to inhibit transcription in cell culture experiments. Previous studies established that transcription can be inhibited with polyamides by targeting the binding sites for essential transcription regulatory proteins in gene promoters in the cell nucleus. We also found that site-specific DNA alkylation by polyamide-chlorambucil conjugates within a coding region of a gene strongly blocks transcription elongation by mammalian RNA polymerase II, both in vitro and in reporter gene transfection experiments in cell culture. We screened a series of polyamide-chlorambucil conjugates with different DNA sequence specificities for effects on morphology and growth characteristics of human colon carcinoma cell lines. We identified a compound that causes cells to arrest in the G2/M stage of the cell cycle, without any apparent cytotoxic effects. This change in growth properties required both the DNAbinding specificity of the polyamide and the alkylator moiety, suggesting that growth arrest is due to the silencing of a set of specific genes by site-specific alkylation. Surprisingly, DNA microarray analysis indicated that only a few genes of about 18,000 genes probed were significantly downregulated by this polyamide, and reverse transcriptase–polymerase chain reaction and Western blotting experiments confirmed that among these genes, a member of the human gene family that encodes histone H4, an essential component of chromatin, is significantly downregulated. This particular gene, the gene for histone H4c, is actively transcribed in various cancer cell lines but is only moderately P Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. transcribed in normal cells and tissues. Downregulation of H4c mRNA by small interfering RNA yielded the same cellular response, providing target validation. The gene for histone H4c contains binding sites for the active polyamide, and DNA alkylation within the coding region of the gene was confirmed in cell culture by using ligation-mediated polymerase chain reaction. Cells treated with this polyamide-chlorambucil conjugate did not grow in soft agar and did not form tumors in nude mice, indicating that polyamide-treated cells are no longer tumorigenic. The compound is active in vivo, blocking tumor growth in mice, without any obvious toxic effects. We extended these studies to various cell lines representing various types of human cancers, including solid tumors of the breast, cervix, lung, pancreas, prostate, and bone and blood cancers, such as leukemias. Our results suggest that polyamide-DNA alkylators may lead to a new class of cancer chemotherapeutic agents. P O LYA M I D E S A S A C T I VAT O R S O F G E N E E X P R E S S I O N In several human diseases, activation of a repressed gene might be useful as a therapeutic approach. One example is the neurodegenerative disease Friedreich’s ataxia, in which gene silencing caused by an unusual DNA structure is the primary cause of the disease. The DNA abnormality found in 98% of patients with Friedreich’s ataxia is the unstable hyperexpansion of a GAA triplet repeat in the first intron of the frataxin gene, which adopts a triplex DNA structure, resulting in decreased transcription and reduced levels of frataxin protein. Frataxin is a mitochondrial protein that functions in iron homeostasis, and decreased levels of frataxin lead to neurodegeneration and cardiomyopathies. We designed pyrrole-imidazole polyamides to target GAA repeats in DNA with high affinity, and we found that these molecules relieved transcription inhibition of the frataxin gene in cell lines and in primary lymphocytes derived from patients with Friedreich’s ataxia. These molecules localize in the cell nucleus, as determined by fluorescence deconvolution microscopy with polyamide-dye conjugates, and most likely reverse repression of the frataxin gene by stabilizing canonical WatsonCrick B-type DNA. Changing the sequence specificities of the molecules abolished their ability to induce frataxin expression. These molecules are a first step toward therapeutic agents for treatment of Friedreich’s ataxia. D N A R E C O G N I T I O N W I T H I N C H R O M AT I N Biochemical and x-ray crystallography studies indicate that nucleosomal DNA is largely available for molec- MOLECULAR BIOLOGY 2005 ular recognition by pyrrole-imidazole polyamides. Polyamide binding sites that are located 80 bp apart on linear DNA lie across the 2 gyres of the DNA superhelix in the nucleosome, forming a supergroove that is unique to the nucleosome. On the basis of this observation, we developed bivalent pyrrole-imidazole polyamide clamps that bind with high specificity across the nucleosomal supergroove. X-ray crystallography studies performed in the laboratory of our collaborator, K. Luger, Colorado State University, indicated that the clamps bind as designed and effectively cross-link the 2 gyres of the DNA superhelix in the nucleosome and stabilize nucleosomal DNA from dissociation. These molecules are useful probes of chromatin structure and dynamics and are tools for regulation of nucleosome mobility during transcription. PUBLICATIONS Beltran, A.C., Dawson, P.E., Gottesfeld, J.M. Role of DNA sequence in the binding specificity of synthetic basic-helix-loop-helix domains. Chembiochem 6:104, 2005. Dickinson, L.A., Burnett, R., Melander, C., Edelson, B.S., Arora, P.S., Dervan, P.B., Gottesfeld, J.M. Arresting cancer proliferation by small-molecule gene regulation. Chem. Biol. 11:1583, 2004. Edayathumangalam, R.S., Weyermann, P., Dervan, P.B., Gottesfeld, J.M., Luger, K. Nucleosomes in solution exist as a mixture of twist-defect states. J. Mol. Biol. 345:103, 2005. Gearhart, M.D., Dickinson, L., Ehley, J., Melander, C., Dervan, P.B., Wright, P.E., Gottesfeld, J.M. Inhibition of DNA binding by human estrogen-related receptor 2 and estrogen receptor with minor groove binding polyamides. Biochemistry 44:4196, 2005. Single-Molecule Conformational Dynamics of Nucleic Acid Enzymes D.P. Millar, M.F. Bailey, G. Pljevaljc̆ić, S. Pond, G. Stengel, N. Tassew, E.J.C. Van der Schans he focus of our research is the assembly and conformational dynamics of nucleic acid–based macromolecular machines. We use single-molecule fluorescence methods to investigate a range of systems, including ribozymes, DNA polymerases, and topoisomerases. Our studies reveal the large structural rearrangements that occur as an integral component of the catalytic mechanism of these enzymes. T RIBOZYMES RNA conformation plays a central role in the mechanism of ribozyme catalysis. The hairpin ribozyme is a small nucleolytic ribozyme that serves as a model sysPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 187 tem for detailed biophysical studies of RNA folding and catalysis. The hairpin ribozyme consists of 2 internal loops, 1 of which contains the scissile phosphodiester bond, displayed on 2 arms of a 4-way multihelix junction. To attain catalytic activity, the ribozyme must fold into a specific conformation in which the 2 loops are docked with each other, forming a network of tertiary hydrogen bonds. We monitor the formation of this docked structure by using fluorescence resonance energy transfer (FRET) and ribozyme constructs labeled with donor and acceptor dyes. By measuring FRET at the level of single ribozyme molecules, we reveal subpopulations of compact and extended conformers that are hidden in conventional experiments. Using this approach, we found that the ribozyme populates an intermediate state in which the 2 loops are in proximity but tertiary interactions have yet to form. This quasi-docked state forms rapidly (submillisecond time scale), but the subsequent formation of tertiary contacts between the loops occurs much more slowly. Surprisingly, the rate of formation of tertiary structure is essentially independent of temperature, indicating that the activation enthalpy is negligible. Hence, the slow tertiary folding is due to an unfavorable entropy change in reaching the transition state. These observations reveal that the tertiary structure of the hairpin ribozyme is formed through a slow conformational search process. This fundamental mechanism of formation of RNA tertiary structure was obscured in most previous folding studies because of the strong propensity of RNA molecules to populate nonnative conformations that act as kinetic traps during the course of folding. D N A P O LY M E R A S E S DNA polymerases are remarkable for their ability to synthesize DNA at rates approaching several hundred base pairs per second while maintaining an extremely low frequency of errors. To elucidate the origin of polymerase fidelity, we are using single-molecule fluorescence methods to examine the dynamic interactions that occur between a DNA polymerase and its DNA and nucleotide substrates. The FRET method is being used to observe conformational transitions of the enzyme-DNA complex that occur during selection and incorporation of an incoming nucleotide substrate. Our results reveal that binding of a correct nucleotide substrate induces a slow conformational change within the polymerase, altering the contacts between the enzyme and the DNA primer/template. This conformational 188 MOLECULAR BIOLOGY 2005 change appears to primarily involve the finger and thumb subdomains of the enzyme. Our studies are providing new insights into the dynamic structural changes responsible for nucleotide recognition and selection by DNA polymerases. Single-pair FRET methods are also being used to monitor the movement of the DNA primer/ template between the separate polymerizing and editing sites of the enzyme. This active-site switching of DNA plays a key role in the proofreading process used to remove misincorporated nucleotides from the newly synthesized DNA. The advantage of single-molecule observations is that they eliminate the need to synchronize a population of molecules, allowing these dynamic processes to be directly observed. TOPOISOMERASES Topoisomerases are enzymes that control the state of DNA supercoiling in the cell. Type I topoisomerases introduce a nick into a strand of DNA and become covalently joined to the cleaved strand. This process allows the other strand to freely swivel around the first, resulting in the relaxation of supercoils within the DNA. The enzyme-DNA connection is then reversed, and the broken strand is rejoined, completing the process of supercoil removal. We are using single-pair FRET methods to observe the DNA-unwinding activity of single type I topoisomerase enzymes in real time. The purpose of these studies is to directly observe DNA rotational motions during supercoil relaxation and to determine whether the same number of supercoils is removed during each enzyme-DNA encounter. PUBLICATIONS Millar, D., Traskelis, M.A., Benkovic, S.J. On the solution structure of the T4 sliding clamp (gp45). Biochemistry 43:12723, 2004. Pljevaljc̆ić, G., Klostermeier, D., Millar, D.P. The tertiary structure of the hairpin ribozyme is formed through a slow conformational search. Biochemistry 44:4870, 2005. Single-Molecule Biophysics A.A. Deniz, S.Y. Berezhna, J.P. Clamme, A.C.M. Ferreon, E.A. Lemke, S. Mukhopadhyay, S. Stanford, P. Zhu e develop and use state-of-the-art singlemolecule fluorescence methods to address key biological questions. Single-molecule and small-ensemble methods offer key advantages over traditional measurements, allowing us to directly observe the behavior of individual subpopulations in mixtures of molecules and to measure kinetics of structural transi- W Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. tions of stochastic processes under equilibrium conditions. We use these methods to study multiple structural states or reaction pathways and stochastic dynamics during the folding and assembly of biomolecules. One major goal is to apply single-molecule methods to studies of protein and RNA folding. Using relatively simple model systems, we are addressing several fundamental questions about folding mechanisms. Partially folded or misfolded protein structures are also thought to play important cellular roles, and these states also can be studied by using single-molecule methods. In this context, we are examining the folding and aggregation of synuclein, a protein implicated in the pathogenesis of Parkinson’s disease and other neurodegenerative diseases. We also continue to use single-pair fluorescence resonance energy transfer (FRET) to study the folding of RNA hairpin ribozymes, in collaboration with D.A. Millar, Department of Molecular Biology. In addition, we are developing a single-molecule fluorescence quenching method that will be useful for measuring distance changes of less than 30 Å in proteins and RNA, a scale at which the resolution of single-pair FRET is low. To better study the folding, assembly, and activity of larger and multicomponent biological complexes, we are developing new multicolor single-molecule FRET methods. As a first step, we developed a diffusion 3-color singlemolecule FRET method by which 2 or more intramolecular or intermolecular distances can be measured simultaneously. In collaboration with J.R. Williamson, Department of Molecular Biology, we are using these novel methods to study the detailed mechanisms of assembly of the bacterial ribosome. The small 30S subunit of the ribosome assembles from a large RNA and 21 small proteins through a complex process that involves several steps of binding and conformational changes. Initially, we are focusing on the conformational properties of small RNA fragments from the 30S subunit and on the interactions of the fragments with their protein partners. These studies are also being extended to the assembly of entire domains of the 30S subunit. Finally, using a combination of high-sensitivity imaging and fluorescence correlation spectroscopy, we are beginning to study the lipid-mediated entry and intracellular delivery pathways of antisense oligodeoxynucleotides and small interfering RNA. An understanding of these mechanisms will be critical to improving the efficiencies of these important genetic tools. MOLECULAR BIOLOGY 2005 PUBLICATIONS Berezhna, S., Schaefer, S., Heintzmann, R., Jahnz, M., Boese, G., Deniz, A.A., Schwille, P. New effects in polynucleotide release from cationic lipid carriers revealed by confocal imaging, fluorescence cross-correlation spectroscopy and single particle tracking. Biochim. Biophys. Acta 1669:193, 2005. Clamme, J.-P., Deniz, A.A. Three-color single-molecule fluorescence resonance energy transfer. Chemphyschem 6:74, 2005. Zhu, P., Clamme, J.-P., Deniz, A.A. Fluorescence quenching by TEMPO: a sub-30 Å single molecule ruler. Biophys. J., in press. Computer Modeling of Proteins and Nucleic Acids D.A. Case, M. Crowley, Q. Cui, P. Dasgupta, F. Dupradeau,* N. Grivel,* R. Lelong,* S. Moon, D. Nguyen, D. Shivakumar, R. Torres, R.C. Walker, L., Yan,* J. Ziegler** * Université Jules Verne, Amiens, France ** Universität Bayreuth, Bayreuth, Germany omputer simulations offer an exciting approach to the study of many aspects of biochemical interactions. We focus primarily on molecular dynamics simulations (in which Newton’s equations of motions are solved numerically) to model the solution behavior of biomacromolecules. Recent applications include detailed analyses of electrostatic interactions in short peptides (folded and unfolded), proteins, and oligonucleotides in solution. In addition, molecular dynamics methods are useful in refining solution structures of proteins by using constraints derived from nuclear magnetic resonance (NMR) spectroscopy, and we continue to explore new methods in this area. Our developments are incorporated into the Amber molecular modeling package, designed for large-scale biomolecular simulations, and into other software, including Nucleic Acid Builder, for developing 3-dimensional models of unusual nucleic acid structures; SHIFTS, for analyzing chemical shifts in proteins and nucleic acids; and RNAmotif, for finding structural motifs in genomic sequence databases. Additional studies on active sites of nitrogenase and other metalloenzymes are described in the report of L. Noodleman, Department of Molecular Biology. C NMR AND THE STRUCTURE AND DYNAMICS OF PROTEINS AND NUCLEIC ACIDS Our overall goal is to extract the maximum amount of information on biomolecular structure and dynamics from NMR experiments. To this end, we are studying the use of direct refinement methods for determining biomolecular structures in solution, going beyond disPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 189 tance constraints to generate closer connections between calculated and observed spectra. We are also using quantum chemistry to study chemical shifts and spinspin coupling constants. Other types of data, such as chemical shift anisotropies, direct dipolar couplings in partially oriented samples, and analysis of cross-correlated relaxation, are also being used to guide structure refinement. In recent structural studies, we focused on minor groove–binding drugs in complex with DNA and on complexes of zinc finger proteins with RNA. NUCLEIC ACID MODELING Another project centers on the development of novel computer methods to construct models of “unusual” nucleic acids that go beyond traditional helical motifs. We are using these methods to study circular DNA, small RNA fragments, and 3- and 4-stranded DNA complexes, including models for recombination sites. We continue to develop efficient computer implementations of continuum solvent methods to allow simplified simulations that do not require a detailed description of the solvent (water) molecules; this approach also provides a useful way to study salt effects. This research is part of a larger effort to develop low-resolution models for nucleic acids that can be extended to much larger structures such as circular DNA, viruses, or models of ribosomal particles. A computer language, NAB, was developed to make it easier to construct and simulate molecular models for complex and often low-resolution problems. The language is being used to study compact and swollen viruses, to analyze curved and circular DNA, and to simulate assembly of ribosomes. D Y N A M I C S A N D E N E R G E T I C S O F N AT I V E A N D N O N N AT I V E S TAT E S O F P R O T E I N S Analysis methods similar to those described for nucleic acids are also being used to estimate thermodynamic properties of “molten globules” and unfolded states of proteins. These studies are an extension of our earlier work on the folding of peptide fragments of proteins. A key feature is the development of computational methods that can be used to model pH and salt dependence of complex conformational transitions, such as unfolding events. A second aspect of this research is a detailed interpretation of NMR results for protein nonnative states through molecular dynamics simulations and the construction of models for molecular motion and disorder. In a parallel effort, we are studying correlated fluctuations about native conformations in a variety of pro- 190 MOLECULAR BIOLOGY 2005 teins, including dihydrofolate reductase, metallo-β-lactamase, binase, and cyclic-dependent kinase, in an effort to make more secure connections between the motions of proteins and the activities of enzymes. All of these modeling activities are based on molecular mechanics force fields, which provide estimates of energies as a function of conformation. We continue to work on improvements in force fields; recently, we focused on adding aspects of electronic polarizability, going beyond the usual fixed-charge models, and on methods for handling arbitrary organic molecules that might be considered potential inhibitors in drug discovery efforts. Overall, the new models should provide a better picture of the noncovalent interactions between peptide groups and the groups’ surroundings, leading ultimately to more faithful simulations. B I O C H E M I C A L S I M U L AT I O N S AT C O N S TA N T p H Like temperature and pressure, the solution pH is an important intensive thermodynamic variable that is commonly varied in experiments and that is used by cells to influence biochemical function. It is now becoming feasible to carry out practical molecular dynamics simulations that mimic the thermodynamics of such experiments, by allowing proton transfer between the system of interest and a hypothetical bath of protons at a given pH. These calculations are demanding, both because the changes in the energetics of charge that occur upon protonation or deprotonation must be accurately modeled and because such simulations must sample both molecular configurations and the large number of protonation states that are possible in a molecule with many acidic or basic sites. This problem is difficult, because almost all biomolecules have multiple sites that can bind or release protons, and these sites are coupled to one another in complex ways. In recent years, however, increases in computational power and new models for estimating the energetics of protonation and deprotonation events have led to serious attempts at simulations that allow the solution pH to be specified as an external variable in a manner that parallels the ways in which temperature or pressure are specified. We recently developed practical methods for estimating ionization probabilities and for allowing the solution pH to be entered as an input variable. Figure 1 shows the results for an acidic group in the protein thioredoxin. The curves show the distribution of energy differences between the protonated and deprotonated forms of the acid or base residue. We can examine the Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 1 . Probability profile for the energy gap (the energy differ- ence between the protonated and deprotonated forms, in kcal/mol) for the side chain of aspartic acid at position 26 in thioredoxin. Values of λ (shown beside the curves) interpolate between the neutral form at λ = 0 and the ionized form at λ = 1. Simple behavior would appear as an inverted parabola; multiple conformations lead to the more complex behavior seen at λ = 0.11. behavior of this variable near the ionized form, corresponding to ordinary pH, or near the neutral, protonated form, at low pH. The results show complex behavior at low pH, which can be analyzed and related to the nature of the acid-base transition under those conditions. These ideas can form the foundation of powerful methods to explore the response of proteins to changes in solvent pH. PUBLICATIONS Baker, N.A., Bashford, D., Case, D.A. Implicit solvent electrostatics in biomolecular simulation. Adv. Macromol. Simul., in press. Beveridge, D.L., Barreiro, G., Byun, K.S., Case, D.A., Cheatham, T.E. III, Dixit, S.B., Giudice, E., Lankas, F., Lavery, R., Maddocks, J.H., Osman, R., Siebert, E., Sklenar, H., Stoll, G., Thayer, K.M., Varnai, P., Young, M.A. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides, 1: research design, informatics, and results on d(CpG) steps. Biophys. J. 87:3799, 2004. Case, D.A., Cheatham, T.E., Darden, T., Gohlke, H., Luo, R., Merz, K.M., Onufriev, A., Simmerling, C., Wang, B., Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem., in press. Mongan, J., Case, D.A. Biomolecular simulations at constant pH. Curr. Opin. Struct. Biol. 15:157, 2005. Mongan, J., Case, D.A., McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 25:2038, 2004. Zhang., Q., Dwyer, T., Tsui, V., Case, D.A., Cho, J., Dervan, P.B., Wemmer, D.E. NMR structure of a cyclic polyamide-DNA complex. J. Am. Chem. Soc. 126:7958, 2004. MOLECULAR BIOLOGY 2005 191 Quantum Chemistry for Intermediates, Reaction Pathways, and Spectroscopy L. Noodleman, D.A. Case, W.-G. Han, F. Himo,* T. Lovell,** T. Liu,*** M.J. Thompson,**** R.A. Torres * Royal Institute of Technology, Stockholm, Sweden ** AstraZeneca R&D, Mölndal, Sweden *** University of Maryland, College Park, Maryland **** Boston University, Boston, Massachusetts F i g . 1 . Proposed model for the active site of class I ribonucleo- tide reductase intermediate X. e use a combination of modern quantum chemistry (density functional theory) and classical electrostatics to describe the energetics, reaction pathways, and spectroscopic properties of enzymes and to analyze systems with novel catalytic, photochemical, or photophysical properties. Critical biosynthetic and regulatory processes may involve catalytic transformations of fairly small molecules or groups by transition-metal centers. The ironmolybdenum cofactor center of nitrogenase catalyzes the multielectron reduction of molecular nitrogen to 2 ammonia molecules plus molecular hydrogen. We are continuing our work on the catalytic cycle of this enzyme, following up on our earlier research on the structure of the MoFe 7S 9X prismane active site, where the central ligand X most likely is nitride. Class I ribonucleotide reductases are aerobic enzymes that catalyze the reduction of ribonucleotides to deoxyribonucleotides, providing the required building blocks for DNA replication and repair. These ribonucleotideto-deoxyribonucleotide reactions occur via a long-range radical (or proton-coupled electron transfer) propagation mechanism initiated by a fairly stable tyrosine radical, “the pilot light.” When this pilot light goes out, the tyrosine radical is regenerated by a high-oxidation-state iron(III)-iron(IV)-oxo enzyme intermediate, called intermediate X. We are using density functional and electrostatics calculations in combination with analysis of Mössbauer, electron nuclear double resonance, and magnetic circular dichroism spectroscopic findings to search for a proper structural and electronic model for intermediate X. On the basis of these studies, we propose that intermediate X contains a di-oxo that bridges the iron(III)-iron(IV) in an asymmetric diamond structure (Fig. 1). In studies with E. Getzoff and M.J. Thompson, Department of Molecular Biology, and D. Bashford, St. W Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Jude Children’s Hospital, Memphis, Tennessee, we are examining the basis for the spectral tuning of the chromophore at the active site of photoactive yellow protein as an example of a light-activated signal transducing protein. In collaborations with K. Hahn, A. Toutchkine, and D. Gremiachinsky, University of North Carolina, Chapel Hill, North Carolina; F. Himo, Royal Institute of Technology, Stockholm, Sweden; and M. Ullmann, University of Bayreuth, Bayreuth, Germany, we examined the optical properties of solvent-dependent fluorescent dyes as prototypes for fluorescent tags that could act as reporters of protein conformational change due to ligand binding. These detailed calculations will be used to improve design strategies for stable and optically useful dyes. Also, with Dr. Bashford’s group, we are studying reaction pathways for the catalytic dephosphorylation of a tyrosine side chain by a low molecular weight protein tyrosine phosphatase. The reaction occurs in 2 distinct steps: first, formation and then hydrolysis of a phosphocysteine intermediate. In a collaboration with K. Janda and T. Dickerson, Department of Chemistry, we used quantum chemical density functional theory methods to examine the mechanism of nornicotine-catalyzed aldol reactions in aqueous solution. Nornicotine is a long-lived nicotine metabolite generated under physiologic conditions in cigarette smokers. This reaction leads to abnormal protein glycation and to covalent modification of steroid drugs, including the prescription corticosteroid prednisone. We are continuing our collaboration with K.B. Sharpless, V.V. Fokin, R. Hilgraf, and V. Rostovtsev, Department of Chemistry, on the catalytic mechanisms used by transition-metal ions in click chemistry, in which metal centers catalyze ring formation from multiply 192 MOLECULAR BIOLOGY 2005 bonded precursors. Our current focus is the mechanism of copper(I) reactions, because copper(I) in water shows great versatility in ligating organic azides and alkynes to form 5-membered heterocycles (triazoles) with wide molecular diversity. On the basis of density function theory calculations, we predict that an unusual 6-membered copper(III) metallocycle intermediate is formed, with only a low barrier to the triazole-copper(I) derivative, leading to the triazole product after proteolysis. PUBLICATIONS Asthagiri, D., Liu, T., Noodleman, L., Van Etten, R.L., Bashford, D. On the role of the conserved aspartate in the hydrolysis of the phosphocysteine intermediate of the low molecular weight tyrosine phosphatase. J. Am. Chem. Soc. 126:12677, 2004. Dickerson, T.J., Lovell, T., Meijler, M.M., Noodleman, L., Janda, K.D. Nornicotine aqueous aldol reactions: synthetic and theoretical investigations into the origins of catalysis. J. Org. Chem. 69:6603, 2004. Himo, F., Lovell, T., Hilgraf, R., Rostovtsev, V.V., Noodleman, L., Sharpless, K.B., Fokin, V.V. Copper(I)-catalyzed synthesis of azoles: DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 127:210, 2005. quantum chemistry to aid in determining the parameters for the models. Calculation of thermodynamic properties requires the development and implementation of new theoretical and computational approaches that connect averages over atomistic descriptions to experimentally measurable thermodynamic and kinetic properties. Interpreting experimental results at more microscopic levels is fueled by the development and investigation of theoretical models of the processes of interest. Massive computational resources are needed to realize these objectives, and this need motivates our efforts aimed at the efficient use of new computer architectures, including large supercomputers, Linux Beowulf clusters, and computational grids. Each of the objectives and techniques mentioned represents an ongoing area of development within our research program in computational biophysics. The following are highlights of a few specific projects. FOLDING, STRUCTURE, AND FUNCTION OF MEMBRANE-BOUND PROTEINS Theoretical and Computational Molecular Biophysics C.L. Brooks III, C. An, R. Armen, I. Borelli, D. Bostick, S.R. Brozell, D. Braun, L. Bu, J. Chen, M.F. Crowley, O. Guvench, R. Hills, W. Im, J. Khandogin, I. Khavrutskii, J. Lee, R. Mannige, M. Michino, H.D. Nguyen, Y.Z. Ohkubo, M. Olson,* S. Patel, D.J. Price, V. Reddy, H.A. Scheraga,** C. Shepard, A. Stoycheva, F.M. Tama, M. Taufer,*** K.A. Taylor,**** I.F. Thorpe, C. Wildman * U.S. Army Medical Research Institute of Infectious Diseases, Fort Detrick, Maryland ** Cornell University, Ithaca, New York *** University of Texas, El Paso, Texas **** Florida State University, Tallahassee, Florida nderstanding the forces that determine the structure of proteins, peptides, nucleic acids, and complexes containing these molecules and the processes by which the structures are adopted is essential to complete our knowledge of the molecular nature of structure and function. To address such questions, we use statistical mechanics, molecular simulation, statistical modeling, and quantum chemistry. Creating atomic-level models to simulate biophysical processes (e.g., folding of a protein or binding of a ligand to a biological receptor) requires (1) the development of potential energy functions that accurately represent the atomic interactions and (2) the use of U Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Folding, insertion, and stability of membrane proteins are directly governed by the unique hydrophilic and hydrophobic environment provided by biological membranes. Modeling this heterogeneous environment is both an obstacle and an essential requisite to experimental and computational studies of the structure and function of membrane proteins. Because of the biological importance and marked presence of membrane proteins in known genomes (i.e., they account for about 30% of all proteins), one aim of modern molecular biophysics should be the development of methods that can be used in experimental studies to understand the structure and function of these systems. We recently developed theoretical methods that enable the exploration of protein insertion and folding in membranes. These methods combine the sampling methods of replica-exchange molecular dynamics with novel generalized Born implicit solvent/implicit membrane continuum electrostatic theories. We recently used de novo folding–membrane association–insertion simulations of a series of peptides (tryptophan-flanked α-helical peptides) designed to explore the concept of hydrophobic mismatch in modulating folding and membrane insertion. Using the simulations, we examined the detailed molecular mechanism of peptide insertion into biological membranes. Our results indicated a common mechanism for the insertion of transmembrane helices of relatively hydrophobic sequences. As illustrated in Figure 1, a peptide MOLECULAR BIOLOGY 2005 becomes associated with the membrane interface, transferring from the aqueous phase, and then helical structure begins to form. The fluctuating helical structure in the interfacial peptide grows until a critical helical length is achieved, and the peptide then inserts via its N-terminal end to form a transmembrane helix. These findings suggest an emerging potential for the de novo investigation of integral membrane peptides and proteins and a mechanism to assist in experimental approaches to characterizing and determining the structure of these important systems. F i g . 1 . Mechanism of membrane association, folding, and inser- tion of a designed membrane peptide. The headgroup regions of the membrane are schematically represented by the parallel plates; the lipid tail–group regions, by the intervening space. Peptides first move from an aqueous environment above the membrane to the interfacial region, where they begin to form helical structure. When the fluctuating helical structure reaches a critical value near 70%–80% helix, the peptide spontaneously inserts from its N-terminal end. 193 large molecular assemblies, including viral capsids, ribosomes, and myosin. In the life cycle of viruses, large-scale reorganization of the protein-protein interfaces of the viral capsid coat is necessary for the functioning of the virus. These motions involve the overall swelling (or shrinking) of the capsid as it reveals (or sequesters) its genome. How such large conformational changes occur is key to understanding and potentially controlling aspects of viral infectivity. Using theoretical methods called elastic network normal mode analysis, we explored putative swelling and shrinking transitions for a number of icosahedral viral capsids of various complexity, from T-numbers of 1 to 13. We discovered a surprisingly similar mechanism for particle expansion and shrinking, despite the significant variation of individual capsid architectures. We examined the collective modes of motion that were energetically easiest to excite, while also directing the conformational change between a swollen (or contracted) icosahedrally symmetric conformation, as observed experimentally. Our calculations (Fig. 2) show that the lowest energy modes that lead to swollen (compressed) states, despite the complexity of the underlying capsid architecture as indicated by the T-number, involves one key mode that produces a uniform deformation of the entire capsid and another that predominately distorts the structures around the 5-fold symmetry axes. Because the mechanical properties, and the global level of deformations necessary for viral functioning, appear to depend solely on the shape of the viral particle, we can hypothesize general mechanisms for a number of viral functions, LARGE-SCALE FUNCTIONAL DYNAMICS IN MOLECULAR ASSEMBLIES Many naturally occurring “machines,” such as ribosomes, myosin, and viruses, require large-scale dynamical motions as a component of their normal functioning. These motions involve the “mechanical” reorganization of major parts of the structure of the machine in response to binding of effectors or to the addition of energy in the form of thermal fluctuations or provided by chemical catalysis. Exploring and understanding the character and nature of such large-scale reorganization of biological machines are ongoing goals in our laboratory. Using theoretical approaches derived from the treatment of mechanoelastic materials, we are constructing theoretical models for the motions of Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . Displacement directions for the swelling of the capsid of the bacteriophage HK97 during maturation from the prohead II state to the head II state as calculated by using elastic network normal mode analysis. The amplitude and direction of motion are indicated by the arrows. The first mode (A) accounts for nearly uniform displacement of all protein units in the capsid, whereas the next lowest energy mode (B) promotes “bulging” around the 5-fold axes of the capsid. 194 MOLECULAR BIOLOGY 2005 from the transfer of genetic material to a host system to the encapsulation of this genetic material in the assembly and maturation of viruses. PUBLICATIONS Chen, J., Brooks, C.L. III, Wright, P.E. Model-free analysis of protein dynamics: assessment of accuracy and model selection protocols based on molecular dynamics simulation. J. Biomol. NMR 29:243, 2004. Chen, J., Im, W., Brooks, C.L. III. Refinement of NMR structures using implicit solvent and advanced sampling techniques. J. Am. Chem. Soc. 126:16038, 2004. Chen, J., Won, H.S., Im, W., Dyson, H.J., Brooks, C.L. III. Generation of nativelike protein structures from limited NMR data, modern force fields and advanced conformational sampling. J. Biomol. NMR 31:59, 2005. Stoycheva, A.D., Brooks, C.L. III, Onuchic, J.N. Gatekeepers in the ribosomal protein S6: thermodynamics, kinetics, and folding pathways revealed by a minimalist protein model. J. Mol. Biol. 340:571, 2004. Tama, F., Brooks, C.L. III. Diversity and identity of mechanical properties of icosahedral viral capsids studied with elastic network normal mode analysis. J. Mol. Biol. 345:299, 2005. Tama, F., Feig, M., Liu, J., Brooks, C.L. III, Taylor, K.A. The requirement for mechanical coupling between head and S2 domains in smooth muscle myosin ATPase regulation and its implications for dimeric motor function. J. Mol. Biol. 345:837, 2005. Tama, F., Miyashita, O., Brooks, C.L. III. Normal mode based flexible fitting of high-resolution structure into low-resolution experimental data from cryo-EM. J. Struct. Biol. 147:315, 2004. Dominy, B.N., Minoux, H., Brooks, C.L. III. An electrostatic basis for the stability of thermophilic proteins. Proteins 57:128, 2004. Taufer, M., Crowley, M., Price, D.J., Chien, A.A., Brooks, C.L. III. Study of a highly accurate and fast protein-ligand docking method based on molecular dynamics. Concurr. Comput. Pract. Exp., in press. Falke, S., Tama, F., Brooks, C.L. III, Gogol, E.P., Fisher, M.T. The 13 Å structure of a chaperonin GroEL-protein substrate complex by cryo-electron microscopy. J. Mol. Biol. 348:219, 2005. Thorpe, I.F., Brooks, C.L. III. The coupling of structural fluctuations to hydride transfer in dihydrofolate reductase. Proteins 57:444, 2004. Feig, M., Brooks, C.L. III. Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr. Opin. Struct. Biol. 14:217, 2004. Feig, M., Im, W., Brooks, C.L. III. Implicit solvation based on generalized Born theory in different dielectric environments. J. Chem. Phys. 120:903, 2004. Feig, M., Onufriev, A., Lee, M.S., Im, W., Case, D.A., Brooks, C.L. III. Performance comparison of generalized Born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 25:265, 2004. Ferrara, P., Gohlke, H., Price, D.J., Klebe, G., Brooks, C.L. III. Assessing scoring functions for protein-ligand interactions. J. Med. Chem. 47:3032, 2004. Guvench, O., Brooks, C.L. III. Efficient approximate all-atom solvent accessible surface area method parameterized for folded and denatured protein conformations. J. Comput. Chem. 25:1005, 2004. Guvench, O., Brooks, C.L. III. Tryptophan side chain electrostatic interactions determine edge-to-face vs parallel-displaced tryptophan side chain geometries in the designed β-hairpin ”trpzip2.” J. Am. Chem. Soc. 127:4668, 2005. Guvench, O., Price, D.J., Brooks, C.L. III. Receptor rigidity and ligand mobility in trypsin-ligand complexes. Proteins 58:407, 2005. Im, W., Brooks, C.L. III. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 102:6771, 2005. Karanicolas, J., Brooks, C.L. III. An evolution of minimalist models for protein folding: from the behavior of protein-like polymers to protein function. Biosilico 2:127, 2004. Mackerell, A.D., Jr., Feig, M., Brooks, C.L. III. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25:1400, 2004. Natrajan, A., Crowley, M., Wilkins-Diehr, N., Humphrey, M.A., Fox, A.D., Grimshaw, A.S., Brooks, C.L. III. Studying protein folding on the Grid: experiences using CHARMM on NPACI resources under Legion. Concurr. Comput. Pract. Exp. 16:385-397, 2004. Patel, S., Brooks, C.L., III. A nonadditive methanol force field: bulk liquid and liquid-vapor interfacial properties via molecular dynamics simulations using a fluctuating charge model. J. Chem. Phys. 122:24508, 2005. Patel, S., Mackerell, A.D., Jr., Brooks, C.L. III. CHARMM fluctuating charge force field for proteins, 2: protein/solvent properties from molecular dynamics simulations using a nonadditive electrostatic model. J. Comput. Chem. 25:1504, 2004. Price, D.J., Brooks, C.L. III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121:10096, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Computation and Visualization in Structural Biology A.J. Olson, D.S. Goodsell, M.F. Sanner, A. Gillet, Y. Hu, R. Huey, C. Li, S. Karnati, W. Lindstrom, G.M. Morris, A. Omelchenko, M. Pique, B. Norledge, R. Rosenstein, D. Stoffler, Y. Zhao n the Molecular Graphics Laboratory, we develop novel computational methods to analyze, understand, and communicate the structure and interactions of complex biomolecular systems. This past year, we showed the effectiveness of 3-dimensional molecular models as a tangible human-computer interface in educational and research settings. Within our component-based visualization environment, we continue to develop methods for predicting biomolecular interactions, analyzing biomolecular structure and function, and presenting the biomolecular world in education and outreach. We have applied these methods to several important systems in human health and welfare. We continue the search for inhibitors of HIV protease to fight the growing problem of drug resistance in HIV disease. We used AutoDock, a suite of programs for predicting bound conformations and binding energies for biomolecular complexes, in the virtual screening of large databases of compounds and ultimately identified new compounds for use in the treatment of cancer. We used methods for predicting protein interactions to probe the mechanism of blood coagulation. I MOLECULAR BIOLOGY 2005 195 TA N G I B L E I N T E R FA C E S F O R S T R U C T U R A L B I O L O G Y We are using the evolving technology of computer autofabrication (“3-dimensional printing”) to produce physical models of complex molecular assemblies (Fig. 1). With this technology, a physical model based on a virtual computer model is built up layer by layer. The great advantage of autofabrication is that nearly any shape can be built; the shape is limited only by the imagination of the researcher and the structural integrity of the building material. We have used 2 technologies: 1 that is much like using a hot glue gun, in which the model is built from layers of molten plastic, and 1 in which gypsum powder and colored binders applied with an ink jet technology are used to create full-color models. F i g . 2 . Top, The augmented reality environment. The user holds F i g . 1 . A sample of the molecular models built by using automated fabrication techniques shows a wide range of molecular representations, scales, and sizes. In collaboration with the Human Interfaces Technology Laboratory at the University of Washington, Seattle, Washington, we developed an augmented reality environment that embeds these 3-dimensional models within the virtual environment of the computer. The goal of this technology is to create a sense of user presence in a computational interaction, combining the intuitive tactile interaction of model manipulation with the rich bioinformatics and visualization tools that are available in the computer environment. As shown in Figure 2, the augmented reality environment tracks the position of the model, displaying a video image of the model and user and overlaying a computer-generated image that is spatially registered with the model as the user manipulates and explores the structure. In tests of the model, high school and college students reported that they experienced a compelling sense of realism of the virtual object and enhanced interaction with the subject matter. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. the model under a FireWire camera. Bottom, A video image of the model is displayed on the computer screen, with an overlaid computer-generated image. Here, the electrostatic potential and field of superoxide dismutase are shown with volume-rendered clouds and small animated arrows. We use the program Python Molecule Viewer to create a diverse range of different representations for both our virtual molecular objects and our tangible models, simplifying integration of the models with the virtual environment. Python Molecule Viewer allows us to combine backbone representation, atomic representations, and surfaces and to incorporate markers for spatial tracking. We are also using computer-aided design and manufacturing methods to design mechanical connectors and magnetic fittings that incorporate aspects of flexibility and interaction into the models. Vision, the visual programming interface, is used to integrate nonmolecular features and properties, such as electrostatics and hydrophobicity, into the virtual and physical environment. C O M P O N E N T - B A S E D V I S U A L I Z AT I O N E N V I R O N M E N T To facilitate the integration and interoperation of computational models and techniques from a wide 196 MOLECULAR BIOLOGY 2005 variety of scientific disciplines, we continue to expand our component-based software environment. The environment is centered on Python, a high-level, object-oriented, interpretive programming language. This approach allows the compartmentalization and reuse of software components. Python provides a powerful “glue” for assembling computational components and, at the same time, a flexible language for the interactive scripting of new applications. We recently added a visual programming environment, Vision, that supports the interactive and visual combination of computational nodes into networks that correspond to algorithms coded at a high level (Fig. 3). Vision provides nonprogrammers an intuitive interface for building networks that describe new computational pipelines and novel visualizations of data. The basic molecular visualization methods of Python Molecule Viewer, a molecular symmetry generator, and a volumerendering method are a few of the currently available nodes, and new nodes are easy to create in the Python language. The combination of the visual programming model and the ability to interactively inspect and edit nodes written in a high-level language creates an unprecedented number of levels at which users can interact with the program. The software tools developed by using our software components have been distributed to more than 10,600 users, with an average of 250 downloads a month during the past year. We released a new version of our software tools in December 2004 that contains a large number of improve- ments and additions. In particular, we streamlined our distribution mechanism and included concurrent versioning system entries that allow users to update the software once it has been installed. We fixed several bugs and added new packages, including mesh decimation algorithms and support for manipulating and visualizing volumetric data. In addition, we increased the number of tests that are run on a nightly basis to more than 2500. MODELING OF FLEXIBILITY In a project funded by the National Institutes of Health, we developed Flexibility Tree, a hierarchical and multiresolution representation of the flexibility of biological macromolecules that can be used in computational simulations. With this software, a user can encode a small subset of a protein’s conformational subspace. After implementing the core infrastructure of Flexibility Tree and integrating it with Python Molecule Viewer and Vision, we are building such trees for molecular systems, including HIV type 1 protease and protein kinases. A number of laboratories around the world have developed software tools for extracting the information that describes how the various parts of proteins move relative to each other. We are now using Flexibility Tree to assess the quality of the decomposition of the protein structure into rigid bodies provided by these tools as well as the accuracy of the motions calculated by using these methods. Early results indicate that when small local perturbations are allowed in addition to the motions predicted by these tools, the Flexibility Tree covers a conformational space that includes both open and closed conformations of our test systems with accuracy sufficient for docking experiments. Our next step will be to design prototype docking tools that can include protein flexibility based on the Flexibility Tree. VIRTUAL SCREENING WITH AUTODOCK F i g . 3 . Vision, a visual programming environment, allows users to build networks of visualization software, creating new computational pipelines and novel visualizations of data. The canvas is shown at the center, where users interactively combine computational nodes. The network shown is a visualization of an electron micrograph reconstruction of a virus, colored by the radial depth and with a sector removed to show the interior structure. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. We have developed new interactive tools to streamline the process of virtual screening in AutoDock. With these tools, users can perform docking experiments to evaluate the binding of a database of molecules with a particular macromolecule of interest. In collaboration with I.A. Wilson, Department of Molecular Biology, we used the method to discover new inhibitors for aminoimidazole carboxamide ribonucleotide transformylase, a target for new cancer chemotherapeutic agents. The diversity set from the National Cancer Institute was screened, and 44 potential candidates were identified. In vitro inhibition assays indicated that 8 of the 44 were soluble compounds, had chemical scaffolds that MOLECULAR BIOLOGY 2005 differed from the general folate template, and caused inhibition when used in micromolar concentrations. Currently, we are optimizing the lead candidates; our goal is to obtain novel nonfolate inhibitors. AutoDock is currently used in more than 3200 academic and commercial laboratories worldwide. We continued development of AutoDock by testing a new empirical free-energy force field. The force field incorporates a charge-based model for evaluation of hydrophobicity and an improved method for evaluating the geometry of hydrogen bonding. The force field was calibrated by using a set of 138 protein complexes of known structure taken from the Ligand Protein Database from the laboratory of C.L. Brooks, Department of Molecular Biology. We anticipate that the revised AutoDock, which incorporates this new force field and methods for selective flexibility in the protein target, will be released in 2005. We also used AutoDock to predict intermolecular interactions in several biological systems. In collaboration with C.F. Barbas, Department of Molecular Biology, we investigated the binding of peptides to the catalytic aldolase antibody 93F3. To explore the large conformational space available to these peptides, we used a divide-and-conquer approach that separates the search space into searchable blocks. In studies with G. Legge, University of Texas, Austin, Texas, we explored the interaction between the cytoplasmic tail of tissue factor and the WW domain of proline isomerase PIN1, focusing on the interaction of several key phosphoserine residues. F I G H T I N G D R U G R E S I S TA N C E I N H I V D I S E A S E We are continuing our work on inhibitors to fight drug resistance in the treatment of AIDS (Fig. 4). In collaboration with K.B. Sharpless and C.-H. Wong, Department of Chemistry, we have focused on the design of inhibitors that assemble within the active 197 site of HIV protease. We showed that the triazole formed in the click chemistry reaction is an effective mimic for the peptide group in traditional inhibitors, forming similar hydrogen-bonding interactions. Currently, we are moving the FightAIDS@Home system from an outside provider to a new server strategy that will be implemented in the Molecular Graphics Laboratory. FightAIDS@Home enlists the worldwide community in a large computational effort to design effective therapeutic agents to fight AIDS. Personal computers are used in the program when the computers are not in use by their owners, providing an enormous, and largely untapped, computational resource. The current goal is to identify inhibitors that are effective against the wild-type virus and against common mutant forms of the virus. The large computational resources provided by FightAIDS@Home enables the screening of large databases of compounds and use of multiple mutant targets, allowing estimation of the potential of a compound to remain effective when viral mutations occur that cause resistance to drugs currently used to treat HIV disease. PREDICTING PROTEIN-PROTEIN INTERACTIONS With the goal of creating a comprehensive tool for predicting protein-protein interactions, we incorporated both SurfDock and AutoDock into the Python programming environment. SurfDock uses a variable-resolution spherical harmonics representation to find candidate orientations, and AutoDock is then used to explore local atomic rearrangements at the interface. We tested the method on a set of 59 protein-protein complexes of known structure and optimized the level of smoothing used in the spherical harmonics approximation of the molecular surfaces. The results of the docking test depended on the force field used to score possible orientations. The best results were obtained with a residue-based pair-wise potential of mean force. V I S U A L M E T H O D S F R O M AT O M S T O C E L L S F i g . 4 . The predicted bound conformation of sanguinarine, a poten- tial lead compound for the development of novel HIV protease inhibitors. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Understanding structural molecular biology is essential to foster progress and critical decision making among students, policy makers, and the general public. In the past year, we continued our longstanding commitment to science education and outreach with a combination of presentations, popular and professional illustrations and animation, 3-dimensional tangible models, and a presence on the Worldwide Web. In these projects, we use the diverse visualization tools developed in the Molecular Graphics Laboratory to disseminate results that range from atomic structure to cellular function. 198 MOLECULAR BIOLOGY 2005 We created a 3-dimensional model that demonstrates viral assembly. The model is composed of pentamers from the structure of poliovirus, with embedded magnets on the interacting faces. When 12 or more of these pentamer models are placed in a closed container and gently shaken, they self-assemble in a matter of seconds to form a spherical capsid. We also continued several regular features that informally present molecular structure and function. The “Molecule of the Month” at the Protein Data Bank (http://www.rcsb.org/pdb) provides an accessible introduction to this central database of biomolecular structure. Each month, a new molecule is presented with a description of its structure, function, and relevance to health and welfare (Fig. 5). Visitors are then given suggestions about how to begin their own exploration of the structures in the data bank. Currently, we are collaborating with T. Herman, Milwaukee School of Engineering, Milwaukee, Wisconsin, to combine material from the “Molecule of the Month” with 3-dimensional models and multimedia tutorials to create educational modules for use at high school and college levels. Other projects include “The Molecular Perspective,” articles in the journal The Oncologist that present structures of interest to clinical oncologists and provide a source of continuing education for physicians, and “Recognition in Action,” a new series in the Journal of Molecular Recognition. Brik, A., Alexandros, J., Lin, Y.-C., Elder, J.H., Olson, A.J., Wlodawer, A., Goodsell, D.S., Wong, C.-H. 1,2,3-Triazole as a peptide surrogate in the rapid synthesis of HIV protease inhibitors. Chembiochem 6:1167, 2005. Gillet, A., Sanner, M., Stoffler, D., Goodsell, D.S., Olson, A.J. Augmented reality with tangible auto-fabricated models for molecular biology applications. In: IEEE Visualization: Proceedings of the Conference on Visualization ’04. IEEE Computer Society, Washington, DC, 2004, p. 235. Gillet, A., Sanner, M., Stoffler, D., Olson, A. Tangible augmented interfaces for structural molecular biology. IEEE Comput. Graph. Appl. 25:13, 2005. Gillet, A., Sanner, M., Stoffler, D., Olson, A. Tangible interfaces for structural molecular biology. Structure (Camb.) 13:483, 2005. Goodsell, D.S. Computational docking of biomolecular complexes with AutoDock. In: Protein-Protein Interactions: A Molecular Cloning Manual, 2nd ed. Golemis, E., Adams, P. (Eds.). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, in press. Goodsell, D.S. The molecular perspective: cyclins. Oncologist 9:592, 2004; Stem Cells 22:1121, 2004. Goodsell, D.S. The molecular perspective: cytochrome c and apoptosis. Oncologist 9:226, 2004; Stem Cells 22:428, 2004. Goodsell, D.S. The molecular perspective: L-asparaginase. Oncologist 10:238, 2005; Stem Cells 23:710, 2005. Goodsell, D.S. The molecular perspective: major histocompatibility complex. Oncologist 10:80, 2005; Stem Cells 23:454, 2005. Goodsell, D.S. The molecular perspective: morphine. Oncologist 9:717, 2004; Stem Cells 23:144, 2005. Goodsell, D.S. The molecular perspective: nicotine and nitrosamines. Oncologist 9:353, 2004; Stem Cells 22:645, 2004. Goodsell, D.S. The molecular perspective: polycyclic aromatic hydrocarbons. Oncologist 9:469, 2004; Stem Cells 22:873, 2004. Goodsell, D.S. Recognition in action: flipping pyrimidine dimers. J. Mol. Recognit. 18:193, 2005. Goodsell, D.S. Representing structural information. In: Current Protocols in Bioinformatics. Baxeranis, A.D., Davison, D.B. (Eds.). Wiley & Sons, Hoboken, NJ, in press. Goodsell, D.S. Visual methods from atoms to cells. Structure (Camb.) 13:347, 2005. Li, C., Xu, L., Wolan, D.W., Wilson, I.A., Olson, A.J. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J. Med. Chem. 47:6681, 2004. Sanner, M.F. A component-based software environment for visualizing large macromolecular assemblies. Structure (Camb.) 13:447, 2005. Sanner, M.F. Using the Python programming language for bioinformatics. In: Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics. Jorde, L.B., Little, P.F.R., Dunn, M.J., et al. (Eds.). Wiley & Sons, Hoboken, NJ, in press. Zhu, X., Tanaka, F., Hu, Y., Heine, A., Fuller, R., Zhong, G., Olson, A.J., Lerner, R.A., Barbas, C.F. III, Wilson, I.A. The origin of enantioselectivity in aldolase antibodies: crystal structure, site-directed mutagenesis, and computational analysis. J. Mol. Biol. 343:1269, 2004. F i g . 5 . Three different types of catalase. Catalase was presented as a Molecule of the Month in 2004 after a request from a high school teacher. PUBLICATIONS Berman, H.M., Ten Eyck, L.F., Goodsell, D.S., Haste, N.M., Kornev, A. Taylor, S.S. The cAMP binding domain: an ancient signaling module. Proc. Natl. Acad. Sci. U. S. A. 102:45, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. MOLECULAR BIOLOGY 2005 Computational Structural Proteomics and Ligand Discovery R. Abagyan, J. An, A. Cheltsov, A. Bordner,* C. Cavasotto,* J. Kovacs, J. Fernandez-Recio,** M. Totrov,* X. Zhang,*** M. Dawson,*** A. McCluskey,**** B. Marsden***** * Molsoft L.L.C., La Jolla, California ** Institut de Recerca Biomèdica, Barcelona, Spain *** Burnham Institute, La Jolla, California **** University of Newcastle, Callaghan, Australia ***** Structural Genomics Consortium, Oxford, England very day about 15 new crystal structures are deposited in the Protein Data Bank. The 30,000 molecular structures in the bank contain rich information about protein function and provide a unique opportunity for rational search for or design of small molecules that can be used as therapeutic agents. We use computational structural proteomics, bioinformatics, molecular mechanics, and cheminformatics to characterize the function of proteins and to design molecular structures. Traditionally, we have focused on accurate docking and screening of small molecules and have used internal coordinate mechanics to predict protein association. In 2004, we focused on improving the information content of evolutionary sequence conservation; predicting and classifying ligand-binding pockets and protein-protein interfaces; improving sequence structure alignments for models by homology; and predicting effects of single-point mutations, loop conformations, and protein association geometry. We also improved protocols for predicting receptor flexibility in ligand docking and applied virtual screening to discover inhibitors of important biomedical targets. E B I O I N F O R M AT I C S A N D P R E D I C T I O N O F P R O T E I N FUNCTION Functional characterization of tens of thousands of proteins is a key computational task. To build 3-dimensional models of structurally uncharacterized protein sequences, we developed a procedure to accurately align those sequences to their Protein Data Bank templates in the areas of weak alignment. The Structural Alignment Database of 1927 alignments was then used to develop improved alignment/threading parameters. Every molecular biologist is confronted with the tasks of discovering and annotating the functions of a protein of interest. A strong evolutionary conservation Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 199 measure in the context of a 3-dimensional model is a powerful source of functional information. However, the currently used measures have a strong dependence on the sequence composition biases of alignments. We developed mathematical formalism that gives a powerful measure of sequence conservation that does not depend on overrepresentation or underrepresentation of certain branches in the alignment. We also used this measure in an improved method to predict novel patches of protein-protein interactions on protein surfaces. Specific association of proteins is a key biological mechanism. However, accurate prediction of interfaces and residues involved in an interaction, often an interaction with an unknown protein partner, is a great challenge for most proteins or domains with known 3-dimensional structure. The preference for any particular interface is subtle because the same surface is also happy to be exposed to water. We attempted to solve that problem by using more meaningful surface properties and more sophisticated numerical methods. Using the optimal docking area method, we showed that with optimized desolvation parameters and an adaptive algorithm of finding the optimal interaction patch, the desolvation signal itself without any other signals can be strong enough. In other studies, we combined a desolvation signal with the improved sequence conservation signal and used the method successfully with a benchmark of 1496 interfaces. PREDICTING PROTEIN STRUCTURE AND A S S O C I AT I O N Predicting partial protein structure or molecular association is a critical task in computational biology and chemistry. This past year we proposed a method to predict both geometry and stabilization energy for single mutations, improved protocols for predicting protein loops, and developed a method to predict largescale protein movements by using simplified protein models represented in internal coordinates. If both partners of a protein complex are known and their “uncomplexed” 3-dimensional models exist or can be built, attempts can be made to predict the association geometry (also called protein docking). In 2004, we used the internal coordinate mechanics docking method successfully in the Critical Assessment of Prediction of Interactions competition, partially because of the improved docking energetics. Although in the first round we predicted only 3 of 7 complexes, in the second and the third rounds, we were correct in 8 of 200 MOLECULAR BIOLOGY 2005 9 tasks. We are working on further improvements of the method. THE CELL POCKETOME Proteins also bind small molecules, the natural substrates or cofactors of the proteins, or specially designed therapeutic agents. Many orphan receptors and uncharacterized surfaces exist. This past year, we further optimized a pocket prediction algorithm and used it successfully on as many as 17,000 pockets from the Protein Data Bank. In this algorithm, a mathematical transformation of the Lennard-Jones potential is used to generate a potential that, contoured at a certain level, specifically locates the potential binding sites with a rather low level of false-positives and false-negatives (Fig. 1). F i g . 1 . Several representatives of a predicted cell pocketome. Using this algorithm, we predicted as many as 96.8% of experimental binding sites at an overlap level of better than 50%. Furthermore, 95% of the predicted sites from the apo receptors were predicted at the same level. We showed that conformational differences between the apo and bound pockets do not dramatically affect the prediction results. The algorithm can be used to predict ligand-binding pockets of uncharacterized protein structures, suggest new allosteric pockets, evaluate the feasibility of inhibition of protein-protein interactions, Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. and prioritize molecular targets. Finally, we collected and classified data for the human cell pocketome, a database of the known and the predicted binding pockets for the human proteome structures. The pocketome can be used for rapid evaluation of possible binding partners of a given chemical compound. We are using the predicted pockets to develop therapeutic molecules that target unexpected binding pockets. Our first result in using such a strategy was obtained in collaboration with D.A. Lomas, University of Cambridge, Cambridge, England; we identified the first small molecules that block the polymerization of the Z mutant of α1-antitrypsin. COMPOUND DOCKING AND VIRTUAL LIGAND SCREENING Small-molecule inhibitors or activators can be discovered rationally by carefully docking them to a target pocket and scoring the result according to the pose and interactions of the small molecule. The virtual screen can be performed against millions of available chemicals or against virtual chemically feasible molecules, and only several dozen computationally selected candidates need to be tested experimentally. We developed and improved different aspects of this strategy and applied it to different drug discovery projects. The docking technology can also help in understanding the structural mechanisms of the actions of small molecules and can be used to rationally design better molecules. Recently, we used the technology to explain the antagonistic effect of an important class of retinoid X receptor antagonists. A major problem in small-molecule docking and screening is protein flexibility and conformational rearrangements of the binding pocket upon ligand binding. This past year we presented several scenarios for incorporating protein flexibility into docking calculations. In some instances, these protocols can be used to simultaneously predict the ligand-binding pose and the pocket rearrangements. PUBLICATIONS Abagyan, R. Problems in computational structural proteomics. In: Structural Proteomics. Sundstrom, M., Norin, M., Edwards, A. (Eds,). CRC Press, Boca Raton, FL, in press. An, J., Totrov, M., Abagyan, R. Comprehensive identification of “druggable” protein ligand binding sites. Genome Inform. Ser. Workshop Genome Inform. 15:31, 2004. An, J., Totrov, M., Abagyan, R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell. Proteomics 4:752, 2005. Bordner, A.J., Abagyan, R. REVCOM: a robust Bayesian method for evolutionary rate estimation. Bioinformatics 21:2315, 2005. MOLECULAR BIOLOGY 2005 201 Bordner, A.J., Abagyan, R. Statistical analysis and prediction of protein-protein interfaces. Proteins 60:353, 2005. Bordner, A.J., Abagyan, R.A. Large-scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins 57:400, 2004. Cavasotto, C.N., Kovacs, J.A., Abagyan, R.A. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 127:9632, 2005. Cavasotto, C.N., Liu, G., James, S.Y., Hobbs, P.D., Peterson, V.J., Bhattacharya, A.A., Kolluri, S.K., Zhang, X.K., Leid, M., Abagyan, R., Liddington, R.C., Dawson, M.I. Determinants of retinoid X receptor transcriptional antagonism. J. Med. Chem. 47:4360, 2004. Cavasotto, C.N., Orry, A.J.W., Abagyan, R.A. The challenge of considering receptor flexibility in ligand docking and virtual screening. Curr. Comput. Aided Drug Des., in press. Cavasotto, C.N., Orry, A.J.W., Abagyan, R. Receptor flexibility in ligand docking. In: Handbook of Theoretical and Computational Nanotechnology. Reith, M., Schommers, W. (Eds.). American Scientific Publishers, Stevenson Ranch, Calif, in press. Fernandez-Recio, J., Abagyan, R., Totrov, M. Improving CAPRI predictions: optimized desolvation for rigid-body docking. Proteins 60:308, 2005. Fernandez-Recio, J., Totrov, M., Skorodumov, C., Abagyan, R. Optimal docking area: a new method for predicting protein-protein interaction sites. Proteins 58:134, 2005. Hill, T.A., Odell, L.R., Quan, A., Abagyan, R., Ferguson, G., Robinson, P.J., McCluskey, A. Long chain amines and long chain ammonium salts as novel inhibitors of dynamin GTPase activity. Bioorg. Med. Chem. Lett. 14:3275, 2004. Kovacs, J.A., Cavasotto, C.N., Abagyan, R.A. Conformational sampling of protein flexibility in generalized coordinates: application to ligand docking. J. Comput. Theor. Nanosci., in press. Marsden, B., Abagyan, R. SAD—a normalized structural alignment database: improving sequence-structure alignments. Bioinformatics 20:2333, 2004. Mass Spectrometry G. Siuzdak, J. Apon, E. Go, K. Harris, R. Lowe, A. Meyers, A. Nordstrom, Z. Shen, C. Smith, G. Tong, S. Trauger, F i g . 1 . A novel nonlinear approach to analyzing mass spectrom- etry data for identification of metabolites. viral proteins. Our results enabled us to examine both local and global viral structure, gaining insight into the dynamic changes of proteins on the viral surface. MASS SPECTROMETRY IN SILICO We are also developing ultra-high-sensitivity approaches in mass spectrometry with a new strategy that involves pulsed laser desorption/ionization from a silylated silicon surface. In desorption/ionization on silicon, silicon is used to capture analytes, and laser radiation is used to vaporize and ionize these molecules. Using this technology, we can analyze a wide range of molecules with unprecedented sensitivity, in the yoctomole range (Fig. 2). W. Uritboonthai, E. Want, W. Webb, C. Wranik M E TA B O L I T E P R O F I L I N G mall molecules ubiquitous in biofluids are now widely used to predict disease states. The inherent advantage of monitoring small molecules rather than proteins is the relative ease of quantitative analysis with mass spectrometry. We are implementing novel mass spectrometry and bioinformatics techniques (Fig. 1) to investigate the metabolite profiles of small molecules as diagnostic indicators of disease. The ultimate goal is to develop analytical and chemical technologies and a data management system to identify and structurally characterize metabolites of physiologic importance. S V I R A L C H A R A C T E R I Z AT I O N We have developed novel methods for characterizing viruses that have applications to whole viruses and Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . Laser desorption/ionization mass spectrometry on struc- tured silylated silicon has sensitivity rivaling that of fluorescence. PUBLICATIONS Bothner, B., Taylor, D., Jun, B., Lee, K.K., Siuzdak, G., Schultz, C.P., Johnson, J.E. Maturation of a tetravirus capsid alters the dynamic properties and creates a metastable complex. Virology 334:17, 2005. 202 MOLECULAR BIOLOGY 2005 Go, E.P., Apon, J.V., Luo, G., Saghatelian, A., Daniels, R.H., Sahi, V., Dubrow, R., Cravatt, B.F., Vertes, A., Siuzdak, G. Desorption/ionization on silicon nanowires. Anal. Chem. 77:1641, 2005. Lacy, E.R., Wang, Y., Post, J., Nourse, A., Webb, W., Mapelli, M., Musacchio, A., Siuzdak, G., Kriwacki, R.W. Molecular basis for the specificity of p27 toward cyclin-dependent kinases that regulate cell division. J. Mol. Biol. 349:764, 2005. Lowe, R., Go, E., Tong, G., Voelcker, N.H., Siuzdak, G. Monitoring EDTA and endogenous metabolite biomarkers from serum with mass spectrometry. Spectroscopy, in press. Saghatelian, A., Trauger, S.A., Want, E., Hawkins, E.G., Siuzdak, G., Cravatt, B.F. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry 43:14332, 2004. Want, E., Cravatt, B.F., Siuzdak, G. The expanding role of mass spectrometry in metabolite profiling and characterization. Chembiochem, in press. Assembly Landscape of the 30S Ribosome J.R. Williamson, F. Agnelli, A. Beck, A. Bunner, A. Carmel, J. Chao, S. Edgcomb, M. Hennig, E. Johnson, D. Kerkow, E. Kompfner, K. Lehmann, H. Reynolds, W. Ridgeway, S.P. Ryder, L.G. Scott, E. Sperling, B. Szymczyna, M. Trevathan he 30S ribosome is 1 of 2 subunits of the 70S ribosome, which is responsible for the synthesis of all proteins in bacterial cells. The 30S ribosome is responsible for decoding the mRNA for protein synthesis. It is composed of a large 16S RNA of approximately 1500 nucleotides and 20 small proteins (S2–S21). The biogenesis of ribosomes consumes approximately half of the energy of the cell in bacteria, and about 20% of the mass of a bacterium is composed of ribosomes. Thus, the assembly of ribosomes must be rapid and efficient. We are using a wide variety of biophysical techniques to study the mechanism of assembly of the 30S ribosome in vitro. We have used nuclear magnetic resonance, x-ray crystallography, isothermal titration calorimetry, single-molecule fluorescence, and transient electric birefringence to probe the details of the mechanism. Pioneering work by Nomura led to the in vitro assembly map for the 30S ribosome: some proteins bind independently to the 16S rRNA, and some require prior binding of other proteins. Using this map as a framework, we used 30S components from Escherichia coli, Thermus thermophilus, and Aquifex aeolicus to do detailed studies. We have constructed an updated and revised assembly map for the 30S subunit (Fig. 1) that contains all of the currently available information about the assembly pathway. T Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . The assembly landscape of the 30S subunit. The confor- mations of the 16S rRNA are represented in the horizontal plane, and the energy of the conformations is the height of the plane. Folding of parallel pathways is indicated by the arrows. The effects of protein binding are schematically illustrated by the 2 successive changes in the landscape. After protein binding (circles), new downhill folding directions are created. All parallel pathways converge on the native 30S conformation at the bottom corner of the landscape. The 30S unit has 3 structural domains, the 5′, central, and 3′, and each of these has one or more primary binding proteins that will bind independently to RNA. This binding is followed by a wave of secondary binding proteins for each domain and a third wave of tertiary binding proteins. Most of the proteins have dependencies solely within their domain; a few of the later binding proteins have interdomain dependencies. The assembly proceeds in a parallel manner, although each domain has a defined hierarchy of binding order. To probe the kinetics of the assembly of the 30S subunit, we developed a novel assay that allows binding of all 20 ribosomal proteins simultaneously. To achieve this simultaneous binding, we initiate assembly of the 30S subunit by combining 16S rRNA with a mixture of all 20 ribosomal proteins uniformly labeled with the stable isotope nitrogen 15. The isotopic label does not perturb the system, but it does result in a mass change of approximately 150 units for each protein. After assembly proceeds for a brief period, we add an excess of unlabeled ribosomal proteins that contain the natural stable isotope nitrogen 14. We can readily determine the amount of the 2 isotopes for each protein by using mass spectrometry. By measuring this fraction as a function of the assembly time, we can monitor the kinetics of all proteins; we term this assay isotope pulsechase kinetics. Using this approach, we did an extensive analysis of the assembly kinetics of the 30S ribosome under a variety of conditions. We systematically varied the concentration of the reaction, the temperature, and the magnesium ion concentration during assembly. Using the temperature dependence of the binding rates, we characterized the activation energy of binding for all of the proteins. We found that the rates of binding are not correlated to the activation energies, and we can MOLECULAR BIOLOGY 2005 monitor many different assembly steps in this complex parallel process. To combine all of the mechanistic information, we have cast the assembly mechanism in terms of an assembly landscape, which has been recently developed in research on protein folding. The assembly landscape of the 30S subunit (Fig. 2) shows the many possible conformations of 16S rRNA in the horizontal plane, and the energy of those conformations is the height of the surface. The 30S final conformation is located at the lower corner of the landscape, but in the absence of ribosomal proteins, it is not the lowest energy conformation. 203 Klostermeier, D., Sears, P., Wong, C.-H., Millar, D.P., Williamson, J.R. A three-fluorophore FRET assay for high-throughput screening of small-molecule inhibitors of ribosome assembly. Nucleic Acids Res. 32:2707, 2004. Lehmann-Blount, K.A., Williamson, J.R. Shape-specific recognition of singlestranded RNA by the GLD-1 STAR domain. J. Mol. Biol. 346:91, 2005. Recht, M.I., Williamson, J.R. RNA tertiary structure and cooperative assembly of a large ribonucleoprotein complex. J. Mol. Biol. 344:395, 2004. Ryder, S.P., Williamson, J.R. Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA 10:1449, 2004. Scott, L.G., Geierstanger, B.H., Williamson, J.R., Hennig, M. Enzymatic synthesis and 19F-NMR studies of 2-fluoroadenine substituted RNA. J. Am. Chem. Soc. 26:11776, 2004. Torres, F.E., Kuhn, P., De Bruyker, D., Bell, A.G., Wolkin, M.V., Peeters, E., Williamson, J.R., Anderson, G.B., Schmitz, G.P., Recht, M.I., Schweizer, S., Scott, L.G., Ho, J.H., Elrod, S.A., Schultz, P.G., Lerner, R.A., Bruce, R.H. Enthalpy arrays. Proc. Natl. Acad. Sci. U. S. A. 101:9517, 2004. Nuclear Magnetic Resonance Studies of RNA and RNA-Ligand Complexes in Solution M. Hennig, N. Kirchner, G.C. Pérez-Alvarado, E.P. Plant,* J.D. Dinman* * University of Maryland, College Park, Maryland iruses constantly threaten human health. Not only are we unable to control infections caused by old enemies such as the influenza virus, but we are continually challenged by new enemies, such as severe acute respiratory syndrome–associated coronavirus (SARS-CoV). Viral mRNAs often contain signals that tell the ribosome to change reading frames during protein synthesis. This recoding event allows viruses to coordinate gene expression from overlapping reading frames such as open reading frames 1a and 1b, which are out-of-frame coding sequences within the SARSCoV genome. Protein 1a is translated directly from open reading frame 1a; the fused polyprotein 1a-1b is produced by programmed –1 ribosomal frameshifting in which the ribosome slips back 1 nucleotide. Like other viral frameshift signals, the SARS-CoV signal contains 2 cis-acting mRNA elements that make up a slippery heptanucleotide site, X XXY YYZ, followed by an adjacent downstream 3′ pseudoknot, a stable mRNA structure. Pseudoknots generally contain 2 stems of doublestranded RNA and 2 or 3 loops of unpaired nucleotides. Our biochemical and solution-state nuclear magnetic resonance studies revealed that the pseudoknot in the SARS-CoV frameshift signal contains 3 stems. Mutagenesis studies indicated that specific sequences V F i g . 1 . The revised assembly map of the 30S subunit. The 16S ribosomal RNA is shown at the top, oriented from 5′ to 3′ direction. Each of the arrows indicates an observed dependency of binding for each ribosomal protein. The primary binding proteins depend solely on interactions with 16S rRNA (top row); the secondary and tertiary binding proteins depend on prior binding of other proteins. The assembly proceeds in many parallel directions, heading downhill on the landscape, and the energy of the RNA is lowered by RNA-folding reactions that create more RNA structure. RNA folding creates the binding sites for the ribosomal proteins, which can then bind, and this binding has an important consequence: new downhill directions are created for more RNA folding. The assembly reaction proceeds by a series of alternating RNA conformational changes and proteinbinding events that eventually result in the complete assembly of the 30S subunit by the convergence of many parallel pathways. PUBLICATIONS Chao, J.A., Williamson, J.R. Joint x-ray and NMR refinement of the yeast L30emRNA complex. Structure (Camb.) 12:1165, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 204 MOLECULAR BIOLOGY 2005 and structures within the pseudoknot are needed for efficient frameshifting, but the exact role of the extra stem in the SARS-CoV frameshifting signal still remains to be determined. Our current results suggest that the 3 stems form a complex globular RNA structure. The elucidation of this structure via high-resolution nuclear magnetic resonance should facilitate the rational development of therapeutic agents designed to interfere with SARS-CoV programmed –1 ribosomal frameshifting and will increase our understanding of how pseudoknots stimulate frameshifting. We continue to develop nuclear magnetic resonance techniques to investigate the structural and functional diversity of RNA. Novel approaches were developed to identify and assign 2′-hydroxyl hydrogens that exchange rapidly with the solvent and thus are difficult to detect in aqueous buffers. The ribose 2′-hydroxyl group distinguishes RNA from DNA and is responsible for differences in conformation, hydration, and thermodynamic stability of RNA and DNA oligonucleotides. This important group lies in the shallow groove of RNA, where it is involved in a network of hydrogen bonds with water molecules stabilizing RNA A-form duplexes. Structural and dynamical information on 2′-hydroxyl protons is essential to understand their respective roles. We provide structural information on 2′-hydroxyl groups in the form of orientational preferences, contradicting the model that the 2′-hydroxyl typically points away from the ribose H-1′ proton. PUBLICATIONS Hennig, M., Fohrer, J., Carlomagno, T. Assignment and NOE analysis of 2′-hydroxyl protons in RNA: implications for stabilization of RNA A-form duplexes. J. Am. Chem. Soc. 127:2028, 2005. Plant, E.P., Pérez-Alvarado, G.C., Jacobs, J.L., Mukhopadhyay, B., Hennig, M., Dinman J.D. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 3:e172, 2005. Components of the Genetic Code in Translation, Cell Biology, and Medicine P. Schimmel, J. Bacher, K. Beebe, Z. Druzina, K. Ewalt, M. Kapoor, E. Merriman, C. Motta, L. Nangle, F. Otero, J. Reader, R. Reddy, M. Swairjo, K. Tamura, E. Tzima, W. Waas, X.-L. Yang T he genetic code was established in the transition from the RNA world to the theater of proteins. The code is an algorithm, matching each Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. amino acid with a nucleotide triplet. The matching of triplets with amino acids occurs through aminoacylation reactions in which enzymes known as aminoacyltRNA synthetases catalyze attachment of each amino acid to its cognate tRNA. Each tRNA, in turn, has an anticodon nucleotide triplet that defines the amino acid–nucleotide triplet relationship of the code. Each amino acid has a single tRNA synthetase. The synthetases are thought to be among the earliest proteins and, as such, essential components of the translation apparatus that established the genetic code and that were present in the last common ancestor of the universal tree of life. As the tree developed and branched into the 3 great kingdoms—archaebacteria, bacteria, and eukaryotes—the enzymes were incorporated into every cell type of every organism. During this long evolutionary period and populating of every cell, the enzymes adopted novel functions while keeping their canonical role as determinates of the genetic code. Related to their central role, the enzymes acquired novel domains enabling them to correct errors of aminoacylation and thereby ensure the stringent accuracy of the code. Unrelated to their canonical activity in translation, their expanded functions include regulation of transcription and translation in bacteria, RNA splicing in fungal organisms, and cytokine signaling in mammalian cells. These novel functions connect translation to other central pathways that control growth, development, and regulation of all cell types. Recently, we have focused on 2 of the expanded functions that have connections to disease and medicine. One function is the editing activity of the synthetases. Mutations in the editing domain of a specific tRNA synthetase cause ambiguity in the genetic code and result in subtle missense substitutions in proteins throughout the organism (Fig. 1). These changes, in turn, cause global changes in protein function. Such changes can, in principle, lead to specific diseases, such as autoimmune disorders. Indeed, specific changes in the phenotypes of mammalian cells in culture occur when an editing-defective synthetase is present. In mammalian cells, tyrosyl- and tryptophanyl-tRNA synthetases are procytokines. When these synthetases are split by alterative splicing or natural proteolysis, specific fragments are released. These fragments are active in signal transduction pathways. For example, T2-TrpRS, a fragment of tryptophanyl-tRNA synthetase, is a potent angiostatic agent. In collaborative experiments with M. Friedlander, Department of Cell Biology, MOLECULAR BIOLOGY 2005 F i g . 1 . Aminoacyl-tRNA synthetases catalyze the attachment of a noncognate amino acid onto tRNA. A distinct hydrolytic second site prevents these substrates from being released for use in protein synthesis. Mutations within the editing site result in the inability to clear noncognate amino acids from the tRNA. These errors in proofreading ultimately lead to incorporation of wrong amino acids into a growing polypeptide. The final result of accumulation of proteins with errors in their primary sequences is cell death. we found that T2-TrpRS arrested angiogenesis in the retina in neonatal mice. The fragment is so effective in arresting angiogenesis that it is now being introduced into a clinical setting for the treatment of blindness caused by macular degeneration. In other research, we are focusing on the usefulness of T2-TrpRS for treatment of highly vascularized tumors. To understand the antiangiogenic activity of T2-TrpRS, we are identifying the cell signaling pathway involved. Recent experiments indicated that vascular endothelial cell cadherin (VE-cadherin), a calcium-dependent adhesion molecule specifically expressed in endothelial cells and essential for normal vascular development, binds directly to T2-TrpRS. This binding, in turn, blocks the proangiogenic activity of vascular endothelial cell growth factor (Fig. 2). Currently, we are examining the mechanism of signaling by T2-TrpRS after it is bound to VEcadherin and the mechanism of export of T2-TrpRS from the cytoplasm to the cell surface. In addition, on the basis of x-ray structures, we proposed a structure-based mechanism for cytokine activation: the structural changes that occur when tryptophanyl- and tyrosyl-tRNA synthetases are split into specific fragments that convert the synthetases to cytokines. In other research, we are investigating the critical steps in the transition from the RNA world to the theater of proteins. Recent findings established a plausible scenario for the selection of L- rather than D -amino Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 205 F i g . 2 . Schematic illustration of proposed model for how T2-TrpRS (T2) interacts with VE-cadherin and blocks signaling pathways for vascular endothelial cell growth vector (VEGF) and its receptor (VEGFR2). acids as the building blocks for proteins in all life forms. Using amino acids activated in a way similar to the way in which modern amino acids are activated, we showed chiral-selective aminoacylation of tRNA-like molecules. We are using x-ray analysis to understand the structural basis of the chiral selectivity. PUBLICATIONS Bacher, J.M., de Crécy-Lagard, V., Schimmel, P. Inhibited cell growth and protein functional changes from an editing-defective tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 102:1697, 2005. Ewalt, K.L., Schimmel, P. Protein biosynthesis: tRNA synthetases. In: Encyclopedia of Biological Chemistry. Lennarz, W.J., Lane, M.D. (Eds.). Academic Press, San Diego, 2004, p. 263. Ewalt, K.L., Yang, X.-L., Otero, F.J., Liu, J., Slike, B., Schimmel, P. Variant of human enzyme sequesters reactive intermediate. Biochemistry 44:4216, 2005. Metzgar, D., Bacher, J.M., Pezo, V., Reader, J., Doring, V., Schimmel, P., Marlière, P., de Crécy-Lagard, V. Acinetobacter sp ADP1: an ideal model organism for genetic analysis and genome engineering. Nucleic Acid Res. 32:5780, 2004. Nordin, B.E., Schimmel, P. Isoleucyl-tRNA synthetases. In: Aminoacyl-tRNA Synthetases. Ibba, M., Francklyn, C., Cusack, S. (Eds.). Landes Bioscience/Eurekah.com, Georgetown, TX, 2005, p. 24. Ribas de Pouplana, L., Musier-Forsyth, K., Schimmel, P. Alanyl-tRNA synthetases. In: Aminoacyl-tRNA Synthetases. Ibba, M., Francklyn, C., Cusack, S. (Eds.). Landes Bioscience/Eurekah.com, Georgetown, TX, 2005, p. 241. Ribas de Pouplana, L., Schimmel, P. Aminoacylations of tRNAs: record-keepers for the genetic code. In: Protein Synthesis and Ribosome Structure: Translating the Genome. Nierhaus, K.H., Wilson, D.N. (Eds.), Wiley-VCH, New York, 2004, p. 169. Schimmel, P. Genetic code. In: McGraw-Hill Encyclopedia of Science and Technology, 10th ed. McGraw-Hill, New York, in press. Schimmel, P., Beebe, K. From the RNA world to the theater of proteins. In: The RNA World, 3rd ed. Gesteland, R.R., Cech, T.R., Atkins, J.F. (Eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, in press. Schimmel, P., Ewalt, K. Translation silenced by fused pair of tRNA synthetases. Cell 119:147, 2004. 206 MOLECULAR BIOLOGY 2005 Schimmel, P., Söll, D. The world of aminoacyl-tRNA synthetases. In: AminoacyltRNA Synthetases. Ibba, M., Francklyn, C., Cusack, S. (Eds.). Landes Bioscience/Eurekah.com, Georgetown, TX, 2005, p. 1. Swairjo, M.A., Schimmel, P. Breaking sieve for steric exclusion of a noncognate amino acid from active site of a tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 102:988, 2005. Tamura, K., Schimmel, P. Non-enzymatic aminoacylation of an RNA minihelix with an aminoacyl phosphate oligonucleotide. Nucleic Acids Symp. Ser. 48:269, 2004. Tang, H.-L., Yeh, L.-S., Chen, N.-K., Ripmaster, T.L., Schimmel, P., Wang, C.-C. Translation of a yeast mitochondrial tRNA synthetase initiated at redundant nonAUG codons. J. Biol. Chem. 279:49656, 2004. Tzima, E., Reader, J.S., Irani-Tehrani, M., Ewalt, K.L., Schwartz, M.A., Schimmel, P. VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J. Biol. Chem. 280:2405, 2005. Mechanisms of RNA Assembly and Catalysis M.J. Fedor, E.M. Calderon, J.W. Cottrell, C.P. Da Costa, J.W. Harger, Y.I. Kuzmin, E.M. Mahen ecent evidence that RNA catalysis participates in regulation of gene expression as well as in RNA processing and protein synthesis underscores the importance of learning the molecular basis of ribozyme activity. The hairpin ribozyme is an especially good model for investigating RNA catalytic mechanisms because of its relative simplicity and the availability of high-resolution structures that provide a framework for evaluating structure-function relationships. This ribozyme catalyzes reversible phosphodiester cleavage through attack of a ribose 2′ oxygen nucleophile on an adjacent phosphorus (Fig. 1). Our goals have been to identify which parts of the ribozyme contribute to catalysis and to understand the chemical basis of this activity. Like all enzymes, hairpin ribozymes combine several strategies to enhance catalytic rate. One important R F i g . 1 . Chemical mechanism of RNA cleavage mediated by the family of small catalytic RNAs that includes the hairpin ribozyme. Cleavage proceeds through an SN2-type mechanism that involves in-line attack of the 2′ oxygen nucleophile on the adjacent phosphorus to form a trigonal bipyramidal transition state in which 5 electronegative oxygen atoms form transient bonds with phosphorus. Breaking of the 5′ oxygen-phosphorus bond generates products with 5′ hydroxyl and 2′,3′-cyclic phosphate termini. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. strategy, which is apparent from crystal structures, is the alignment of nucleophilic and leaving-group oxygens in the optimal orientation for an SN2-type nucleophilic attack. Biochemical and structural studies also implicate 2 active-site nucleobases, guanine 8 and adenine 38, in catalytic chemistry; the N-1 ring nitrogen of guanine 8 is located near the 2′ oxygen that acts as the nucleophile during cleavage, and the N-1 ring nitrogen of adenine 38 is located near the 5′ oxygen leaving group. Ribonuclease A is a protein enzyme that catalyzes the same chemical reaction as hairpin ribozyme cleavage and has 2 active-site histidines that occupy positions similar to those of guanine 8 and adenine 38. Ribonuclease A provides a textbook example of concerted general acid-base catalysis, and the similarity between hairpin ribozyme and ribonuclease A activesite structures led to the idea that guanine 8 and adenine 38 might serve as general acid and base catalysts as the histidines of ribonuclease A do. The activity of the hairpin ribozyme increases with increasing pH, consistent with the notion that activity depends on the availability of guanine 8, in its unprotonated form, to accept a proton to activate the 2′ hydroxyl nucleophile as proposed in the general acid-base catalysis model. However, a ribozyme variant in which guanine 8 is replaced by an abasic residue has the same pH dependence as an unmodified ribozyme, suggesting that the pH transition in activity does not involve guanine 8. These data support an alternative model in which the protonated form of guanine 8 donates hydrogen bonds that provide electrostatic stabilization as negative charge develops in the transition state (Fig. 2). Replacing adenine 38 with an abasic residue, on the other hand, does eliminate this pH-dependent transition, evidence that the protonation state of adenine 38 is important for activity. The activity that is lost when adenine 38 or guanine 8 is replaced by abasic residues can be rescued by certain nucleobases provided in solution. The molecules that can rescue activity all have planar structures and an amidine group, that is, an amino group in α-position to a ring nitrogen. The same feature is shared with the Watson-Crick face of the missing adenine and guanine, suggesting that chemical rescue occurs through binding of exogenous nucleobases in the cavity left by an abasic substitution. Purines that lack an amidine group can inhibit chemical rescue, presumably by competing with rescuing nucleobases for binding in the cav- MOLECULAR BIOLOGY 2005 207 Directed Evolution of Nucleic Acid Enzymes G.F. Joyce, T.A. Jackson, G.C. Johns, H.R. Kalhor, C.-Y. Lai, M. Oberhuber, B.M. Paegel, G.G. Springsteen, S.B. Voytek ll life known to exist on Earth today is based on DNA genomes and protein enzymes, but strong evidence indicates that it was preceded by a simpler form of life based on RNA. This earlier era is referred to as the “RNA world.” During that time, genetic information resided in the sequence of RNA molecules, and phenotype was derived from the catalytic behavior of RNA. By studying the properties of RNA in the laboratory, especially with regard to the evolution of catalytic function, we can gain insight into the RNA world. In addition, we can develop novel nucleic acid enzymes that have applications in biology and medicine. A F i g . 2 . Results of mechanistic studies of the hairpin ribozyme are consistent with 2 models in which the functional form of adenine 38 is either protonated or unprotonated. In the first model (A), protonated adenine 38 would act as a general acid by donating a proton to the 5′ oxygen, acting in concert with hydroxide ion that activates the 2′ oxygen nucleophile during cleavage, and unpro- tonated adenine 38 would act as a general base to activate the 5′ oxygen nucleophile during ligation. In the second model (B), unprotonated adenine 38 accepts a hydrogen bond from the 5′ hydroxyl nucleophile during ligation and accepts a hydrogen bond from a protonated bridging 5′ oxygen during cleavage, providing electrostatic stabilization to developing negative charge. In both models, the amidine group of guanine 8, in its protonated form, donates hydrogen bonds to the 2′ and phosphoryl oxygens that stabilize the negative charge that develops in the transition state and that position reactive groups in the orientation appropriate for an SN2 in-line nucleophilic attack. ity left by the abasic substitution. Thus, rescue does not occur through binding alone, and amidine functional groups must form specific stabilizing interactions with the transition state. The pH dependence of chemical rescue of ribozymes lacking adenine 38 changes according to the intrinsic basicity of the rescuing nucleobase. These and other results are consistent with 2 models of the hairpin ribozyme catalytic mechanism in which adenine 38 contributes either general acid-base catalysis (Fig. 2A) or electrostatic stabilization of negative charge that develops as 5 electronegative oxygen atoms form transient bonds with phosphorus in the transition state (Fig. 2B). PUBLICATIONS Fedor, M.J., Williamson, J.R. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell Biol. 6:399, 2005. Kuzmin, Y.I, Da Costa, C.P., Cottrell, J.W., Fedor, M.J. Role of an active site adenine in hairpin ribozyme catalysis. J. Mol. Biol. 349:989, 2005. Mahen, E.M., Harger, J.W., Calderon, E.M., Fedor, M.J. Kinetics and thermodynamics make different contributions to RNA folding in vitro and in yeast. Mol. Cell 19:27, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. S Y N T H E S I S A N D D E R I VAT I Z AT I O N O F R I B O S E Ribose, the sugar component of RNA, is a minor component among the many products of the condensation of formaldehyde. In addition, ribose is more reactive than most other sugars and degrades more rapidly than they do. Thus, it is difficult to understand why ribose is included in the genetic material. We exploited the greater reactivity of ribose by allowing it to react preferentially with cyanamide to form a stable product. This product crystallized spontaneously in aqueous solution under a broad range of conditions; the corresponding cyanamides derived from other sugars did not. Furthermore, the ribose-cyanamide crystals reacted with cyanoacetylene to form cytosine α-nucleoside in nearly quantitative yield. The RNA-catalyzed synthesis of ribose from simple starting materials would have been an essential reaction in the RNA world. We approached this problem by examining the ability of a nucleic acid template to direct the synthesis of ribose from 2 aldehyde-bearing oligonucleotides, one with glyceraldehyde at its 3′ end and the other with glycoaldehyde at its 5′ end. The 2 oligonucleotides were allowed to bind at adjacent positions along a complementary template, resulting in an aldol reaction that gave rise to pentose sugars (Fig. 1). No reaction was detected in the absence of the template. Adding lysine to the mixture increased the reaction rate substantially. This reaction will be used as the basis for in vitro evolution experiments to obtain RNAs that catalyze the formation of ribose. 208 MOLECULAR BIOLOGY 2005 F i g . 1 . RNA-directed synthesis of pentose sugars via aldol condensation. Two oligonucleotides, one with glyceraldehyde at its 3′ end (S1) and the other with glycoaldehyde at its 5′ end (S2), are joined in the presence of a complementary template to form a pentose-linked product. C R O S S - R E P L I C AT I N G R N A E N Z Y M E S The central process of the RNA world was the RNAcatalyzed replication of RNA. We previously developed an RNA enzyme, termed the R3C ligase, that catalyzes the template-directed joining of 2 RNA molecules. This enzyme was converted to a format that allows it to produce additional copies of itself through the joining of 2 component subunits. The copies in turn give rise to additional copies, resulting in an exponential increase in the number of enzyme molecules over time. We further modified the reaction system so that it would operate cross-catalytically, whereby 2 RNA enzymes catalyze each other ’s synthesis from a total of 4 substrates (Fig. 2). The newly formed copies of each enzyme give rise to additional copies of the cross-catalytic products, and the rate of formation of both enzymes increases during the course of the reaction. Currently, the crossreplicating system operates with a highly restricted set of RNA sequences, but it provides an opportunity for developing more efficient and more complex networks of replicating RNAs. CONTINUOUS EVOLUTION OF RNA ENZYMES Previously, we developed a powerful method for the in vitro evolution of RNA enzymes that catalyze the joining of RNA molecules. Rather than manipulating the RNAs through successive steps of reaction, selection, and amplification, we devised a way to have these steps occur continuously within a common reaction vessel. Evolution can be carried out indefinitely by a serial transfer procedure, whereby a small part of a completed Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 2 . Cross-catalytic replication of RNA enzymes. The enzyme E binds the substrates S1′ and S2′ and catalyzes their joining to form the enzyme E′. Similarly, the enzyme E′ binds and joins the substrates S1 and S2 to form the enzyme E. reaction mixture is transferred to a new reaction vessel that contains a fresh supply of substrates and the other components necessary for selective amplification. During the past year, we began 3 new lines of investigation involving continuous in vitro evolution. First, we modified the system so that an increased frequency of random mutations would occur during amplification. This modification allows us to generate and exploit genetic diversity within the system, providing a more realistic model of biological evolution. Second, using either 2 distinct variants of 1 enzyme or 2 different enzymes, we sought to evolve 2 different RNA enzymes within a common environment. These evolved enzymes will be used to study competition and cooperation in the context of RNA-based evolution. Third, we implemented a novel microfluidic system for continuous in vitro evolution. In this system, the population of enzymes is confined to a microfluidic circuit within a fabricated glass wafer that contains a middle layer of an elastomeric material that functions as control valves. The concentration of RNA is monitored by using a confocal fluorescence microscope, and serial transfer is triggered automatically whenever the population size reaches a predetermined threshold. The MOLECULAR BIOLOGY 2005 209 microfluidic system makes it possible to conduct thousands of generations of in vitro evolution in a highly precise manner with little intervention by the experimenter. PUBLICATIONS Johns, G.C., Joyce, G.F. The promise and peril of continuous in vitro evolution. J. Mol. Evol. 61:253, 2005. Joyce, G.F., Orgel, L.E. Progress toward understanding the origin of the RNA world. In: The RNA World, 3rd ed. Gesteland, R.F., Cech, T.R., Atkins, J.F. (Eds.). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, in press. Kim, D.-E., Joyce, G.F. Cross-catalytic replication of an RNA ligase ribozyme. Chem. Biol. 11:1505, 2004. Paul, N., Joyce, G.F. Minimal self-replicating systems. Curr. Opin. Chem. Biol. 8:634, 2004. Springsteen, G., Joyce, G.F. Selective derivatization and sequestration of ribose from a prebiotic mix. J. Am. Chem. Soc. 126:9578, 2004. Studies at the Interface of Molecular Biology, Chemistry, and Medicine C.F. Barbas III, B.A. Gonzalez, L. Asawapornmongkul, D.B. Ramachary, S. Eberhardy, R. Fuller, R. Gordley, J. Guo, B. Henriksen, C. Lund, J. Mandell, S. Mitsumori, R. Mobini, N.S. Chowdari, M. Popkov, D. Steiner, J. Suri, F. Tanaka, U. Tschulena, Y. Ye, Y. Yuan, G. Zhong e are concerned with problems in molecular biology, chemistry, and medicine. Many of our studies involve learning or improving on Nature’s strategies to prepare novel molecules that perform specific functional tasks, such as regulating a gene, destroying cancer, or catalyzing a reaction with enzymelike efficiency. We hope to apply these novel insights, technologies, methods, and products to provide solutions to human diseases, including cancer, HIV disease, and genetic diseases. W D I R E C T I N G T H E E V O L U T I O N O F C ATA LY T I C F U N C T I O N Using our concept of reactive immunization, we have developed antibodies that catalyze aldol as well as retro-aldol reactions of a wide variety of molecules. The catalytic proficiency of the best of these antibodies is almost 1014, a value 1000 times that of the best catalytic antibodies reported to date and overall the best of any synthetic protein catalyst. We have shown the efficient asymmetric synthesis and resolution of a variety of molecules, including tertiary and fluorinated aldols, and have used these chiral synthons to synthesize natural products (Fig. 1). The results Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 1 . A variety of compounds synthesized with the world’s first commercially available catalytic antibody, 38C2, produced at Scripps Research. highlight the potential synthetic usefulness of catalytic antibodies as artificial enzymes in addressing problems in organic chemistry that are not solved by using natural enzymes or more traditional synthetic methods. To further evolve these catalytic antibodies, we are developing genetic selection methods. Other advances in this area include the development of the first peptide aldolase enzymes. Using both design and selection, we created small peptide catalysts that recapitulate many of the kinetic features of large protein catalysts. With these smaller enzymes, we can address how the size of natural proteins is related to catalytic efficiency. O R G A N O C ATA LY S I S : A B I O O R G A N I C A P P R O A C H T O C ATA LY T I C A S Y M M E T R I C C A R B O N - C A R B O N BOND–FORMING REACTIONS To further explore the principles of catalysis, we are studying amine catalysis as a function of catalytic scaffold. Using insights garnered from our studies of aldolase antibodies, we determined the efficacy of simple chiral amines and amino acids for catalysis of aldol and related imine and enamine chemistries such as Michael, Mannich, Knoevenagel, and Diels-Alder reactions (Fig. 2). Although aldolase antibodies are superior in terms of the kinetic parameters, these more simple catalysts are enabling us to quantify the importance of pocket sequestration in catalysis. Furthermore, many of these catalysts are cheap, environmentally friendly, and practical for large-scale synthesis. With this approach, we showed the scope 210 MOLECULAR BIOLOGY 2005 F i g . 2 . L -Proline and other organocatalysts developed for a variety of catalytic asymmetric syntheses via aldol, Michael, Mannich, Diels-Alder, and Knoevenagel reactions provide access to important classes of compounds. These catalysts make reactions that were once complex multistep reactions, simple 1-step reactions. A wide variety of medicinally important products can be assembled by using the Mannich reaction manifold alone. and usefulness of the first efficient amine catalysts of direct asymmetric aldol, Mannich, Diels-Alder, and Michael reactions. The organocatalyst approach is a direct outcome of our studies of catalytic antibodies and provides an effective alternative to organometallic reactions that use severe reaction conditions and oftentoxic catalysts. We think that our discovery that the simple naturally occurring amino acids such as L-proline and other amines can effectively catalyze a variety of enantioselective intermolecular reactions will change the way many reactions will be performed. Furthermore, these catalysts are functional in related ketone addition reactions such as Mannich- and Michael-type reactions. As a testament to the mild nature of this approach, we developed the first catalytic asymmetric aldol, Mannich, Michael, and fluorination reactions involving aldehydes as nucleophiles. Previously, such reactions were considered out of the reach of traditional synthetic methods. In an extension of these concepts, we invented a variety of novel multicomponent or asymmetric assembly reactions (Fig. 3). Our finding that a variety of optically active amino acids can be synthesized with proline catalysis in which an L-amino acid begets other L-amino acids suggests that this route may have been used in prebiotic syntheses of optically active amino acids. In addition, we showed that our strategy can be used to synthesize carbohydrates directly, thereby providing a provocative prebiotic route to the sugars essential for life. Unlike most catalysts obtained via traditional approaches, our catalysts are environmentally safe and are available in both enantiomeric forms. The reactions do not require inert conditions or heavy metals and can be performed at room temperature without preactivation of the donor substrates. Because amines can Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 3 . A few recently developed catalytic asymmetric assembly reactions. In these reactions, designed small organic molecules are used to synthesize complex molecules. act as catalysts via both nucleophilic (enamine based) and electrophilic (iminium based) activation, they have great potential in catalytic asymmetric synthesis. THERAPEUTIC ANTIBODIES, IN AND OUT OF CELLS We developed the first human antibody phage display libraries and the first synthetic antibodies and methods for the in vitro evolution of antibody affinity. The ability to manipulate large libraries of human antibodies and to evolve such antibodies in the laboratory provides tremendous opportunities to develop new medicines. Laboratories and pharmaceutical companies around the world now apply the phage display technology that we developed for antibody Fab fragments. In our laboratory, we are targeting cancer and HIV disease. One of our antibodies, IgG1-b12, protects animals against primary challenge with HIV type 1 (HIV-1) and has been further studied by many other researchers. We improved this antibody by developing in vitro evolution strategies that enhanced its neutralization activity. By coupling laboratory-evolved antibodies with potent toxins, we showed that immunotoxins can effectively kill infected cells. We are also developing genetic methods to halt HIV by using gene therapy. We created unique human MOLECULAR BIOLOGY 2005 211 antibodies that can be expressed inside human cells to make the cells resistant to HIV infection. In the future, these antibodies might be delivered to the stem cells of patients infected with HIV-1, allowing the development of a disease-free immune system that would preclude the intense regimen of antiviral drugs now required to treat HIV disease. Using our increased understanding of antibody-antigen interactions, we extended our efforts in cancer therapy and developed rapid methods for creating human antibodies from antibodies derived from other species. We produced human antibodies that should enable us to selectively starve a variety of cancers by inhibiting angiogenesis and antibodies that will be used to deliver radionuclides to colon cancers to destroy the tumors. We hope that some of these antibodies will be used in clinical trials done by our collaborators at the SloanKettering Cancer Center in New York City. On the basis of our studies on HIV-1, we used intracellular expression of antibodies directed against angiogenic receptors to create a new gene-based approach to cancer. We are determining if this new approach can be applied in vivo to halt tumor growth. Our preliminary results indicate that this type of gene therapy can be successfully applied to the treatment of cancer. peutically relevant concentrations. The efficacy of this approach has been shown in in vivo models of cancer. Currently, we are developing more potent drugs and novel antibodies that will allow us to target breast, colon, and prostate cancer as well as cells infected with HIV-1. On the basis of our preliminary findings, we think that our approach can become a key tool in selective chemotherapeutic strategies. To see a movie illustrating this approach, visit http://www.scripps.edu/mb/barbas/index.html. T H E R A P E U T I C A P P L I C AT I O N S O F C ATA LY T I C ADAPTOR IMMUNOTHERAPY: THE ADVENT OF ANTIBODIES The development of highly efficient catalytic antibodies opens the door to many practical applications. One of the most fascinating is the use of such antibodies in human therapy. We think that use of this strategy can improve chemotherapeutic approaches to diseases such as cancer and AIDS. Chemotherapeutic regimens are typically limited by nonspecific toxic effects. To address this problem, we developed a novel and broadly applicable drug-masking chemistry that operates in conjunction with our unique broad-scope catalytic antibodies. This masking chemistry is applicable to a wide range of drugs because it is compatible with virtually any heteroatom. We showed that generic drug-masking groups can be selectively removed by sequential retro-aldol–retro-Michael reactions catalyzed by antibody 38C2 (Fig. 4). This reaction cascade is not catalyzed by any known natural enzyme. Application of this masking chemistry to the anticancer drugs doxorubicin, camptothecin, and etoposide produced prodrugs with substantially reduced toxicity. These prodrugs are selectively unmasked by the catalytic antibody when the antibody is applied at theraPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 4 . Targeting cancer and HIV with prodrugs activated by cat- alytic antibodies. A bifunctional antibody is shown targeting a cancer cell for destruction. A nontoxic analog of doxorubicin, prodoxorubicin, is being activated by an aldolase antibody to the toxic form of the drug. CHEMOBODIES We think that combining the chemical diversity of small synthetic molecules with the immunologic characteristics of antibody molecules will lead to therapeutic agents with superior properties. Therefore, we developed a conceptually new device that equips small synthetic molecules with both the immunologic effector functions and the long serum half-life of a generic antibody molecule. For a prototype, we developed a targeting device based on the formation of a covalent bond of defined stoichiometry between (1) a 1,3-diketone derivative of an arginine–glycine–aspartic acid peptidomimetic that targets the integrins αvβ3 and αvβ5 and (2) the reactive lysine of aldolase antibody 38C2. The resulting complex spontaneously assembled in vitro and in vivo, selectively retargeted antibody 38C2 to the surface of cells expressing integrins αvβ3 and αvβ5, dramatically increased the circulatory half-life of the peptidomimetic, and effectively reduced tumor growth in animal models of human Kaposi sarcoma and colon cancer (Fig. 5). These studies have been extended to melanoma. ZINC FINGER GENE SWITCHES The solution to many diseases might be simply turning genes on or off in a selective way. In order to pro- 212 MOLECULAR BIOLOGY 2005 F i g . 5 . Adaptor Immunotherapy dramatically slows tumor growth. A variety of cancer xenografts have been effectively treated with chemobodies, a combination of small-molecule drugs and antibodies. Chemobodies have characteristics that can be superior to those of either the small molecule or the antibody alone. duce switches that can turn genes on or off, we are studying molecular recognition of DNA by zinc finger proteins and methods of creating novel zinc finger DNAbinding proteins (Fig. 6). Because of their modularity and well-defined structural features, zinc finger proteins are particularly well suited for use as DNA-binding proteins. Each finger forms an independently folded domain that typically recognizes 3 nucleotides of DNA. We showed that proteins can be selected or designed that contain zinc fingers that recognize novel DNA sequences. These studies are aiding the elucidation of rules for sequence-specific recognition within this family of proteins. We selected and designed specific zinc finger domains that will constitute an alphabet of 64 domains that will allow any DNA sequence to be bound selectively. The prospects for this “second genetic code” are fascinating and should have a major impact on basic and applied biology. We showed the potential of this approach in multiple mammalian and plant cell lines and in whole organisms. With the use of characterized modular zinc finger domains, polydactyl proteins capable of recognizing an 18-nucleotide site can be rapidly constructed. Our results suggest that zinc finger proteins might be useful as genetic regulators for a variety of human ailments and provide the basis for a new strategy in gene therapy. Our goal is to develop this class of therapeutic proteins to inhibit or enhance the synthesis of proteins, providing a direct strategy for fighting diseases of either somatic or viral origin. We are also developing proteins that will inhibit the growth of tumors and others that will inhibit the expression of a protein known as CCR5, which is a key to infection of human cells by HIV-1. We developed an HIV-1–targeting transcription factor that strongly suppresses HIV-1 replication. Genetic diseases such as sickle cell anemia are also being targeted. Using a library of transcription factors, we developed a strategy that effectively allows us to turn on and turn off every gene in the genome. With this powerful new strategy, we can quickly regulate a target gene or discover other genes that have a key role in disease. In the future, we hope to use novel DNA-modifying enzymes directed by zinc fingers to manipulate chromosomes themselves. PUBLICATIONS Amir, R.J., Popkov, M., Lerner, R.A., Barbas, C.F. III, Shabat, D. Prodrug activation gated by a molecular “OR” logic trigger. Angew. Chem. Int. Ed. 44:4378, 2005. Betancort, J.M., Sakthivel, K., Thayumanavan, R., Tanaka, F., Barbas, C.F. III. Catalytic direct asymmetric Michael reactions: addition of unmodified ketone and aldehyde donors to alkylidene malonates and nitro olefins. Synthesis 1509, 2004, Issue 9. Blancafort, P., Segal, D.J., Barbas, C.F. III. Designing transcription factor architectures for drug discovery. Mol. Pharmacol. Rev. 66:1361, 2004. F i g . 6 . A designed polydactyl zinc finger binds 18 bp of DNA. A single zinc finger domain is highlighted. With this direct approach, we can construct more than a billion gene switches and use the switches to specifically turn genes on or off in multiple organisms. With further elaboration of the approach, every gene in the genome can be either upregulated or downregulated, providing a new approach to probe gene function across the genome. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Chen, E.I., Florens, L., Axelrod, F.T., Monosov, E., Barbas, C.F. III, Yates, J.R. III, Felding-Habermann, B., Smith, J.W. Maspin alters the carcinoma proteome. FASEB J. 19:1123, 2005. Chowdari, N.S., Barbas, C.F. III. Total synthesis of LFA-1 antagonist BIRT-377 via organocatalytic asymmetric construction of a quaternary stereocenter. Org. Lett. 7:867, 2005. Chowdari, N.S., Suri, J.T., Barbas, C.F. III. Asymmetric synthesis of quaternary α- and β-amino acids and β-lactams via proline catalyzed Mannich reactions with branched aldehyde donors. Org. Lett. 6:2507, 2004. MOLECULAR BIOLOGY 2005 213 Crotty, J.W., Etzkorn, C., Barbas, C.F. III, Segal, D.J., Horton, N.C. Crystallographic analysis of Aart, a designed six-finger zinc finger peptide, bound to DNA. Acta Crystallogr. F61:573, 2005. Tanaka, F., Mase, N., Barbas, C.F. III. Determination of cysteine concentration by fluorescence increase: reaction of cysteine with a fluorogenic aldehyde. Chem. Commun. (Camb.) 1762, 2004, Issue 15. Gräslund, T., Li, X., Popkov, M., Barbas, C.F. III. Exploring strategies for the design of artificial transcription factors: targeting sites proximal to known regulatory regions for the induction of γ-globin expression and the treatment of sickle cell disease. J. Biol. Chem. 280:3707, 2005. Thayumanavan, R., Tanaka, F., Barbas, C.F. III. Direct organocatalytic asymmetric aldol reactions of α-amino aldehydes: expedient synthesis of highly enantiomerically enriched anti-β-hydroxy-α-amino acids. Org. Lett. 6:3541, 2004. Haba, K., Popkov, M., Shamis, M., Lerner, R.A., Barbas, C.F. III, Shabat, D. Single-triggered trimeric prodrugs, Angew. Chem. Int. Ed. 44:716, 2005. Jendreyko, N., Popkov, M., Rader, C., Barbas, C.F. III. Phenotypic knockout of VEGF-R2 and Tie-2 with an intradiabody reduces tumor growth and angiogenesis in vivo. Proc. Natl. Acad. Sci. U. S. A. 102:8293, 2005. Zhong, G., Fan, J., Barbas, C.F. III. Amino alcohol catalyzed direct asymmetric aldol reactions: enantioselective synthesis of anti-α-fluoro-β-hydroxy ketones. Tetrahedron Lett. 45:5681, 2004. Zhu, X., Tanaka, F., Hu, Y., Heine, A., Fuller, R., Zhong, G., Olson, A.J., Lerner, R.A., Barbas, C.F. III, Wilson, I.A. The origin of enantioselectivity in aldolase antibodies: crystal structure, site-directed mutagenesis, and computational analysis. J. Mol. Biol. 343:1269, 2004. Li, L.-S., Rader, C., Matsushita, M., Das, S., Barbas, C.F, III, Lerner, R.A., Sinha, S.C. Chemical adaptor immunotherapy: design, synthesis, and evaluation of novel integrin-targeting devices. J. Med. Chem. 47:5630, 2004. Magnenat, L., Blancafort, P., Barbas, C.F. III. In vivo selection of combinatorial libraries and designed affinity maturation of polydactyl zinc finger transcription factors for ICAM-1 provides new insights into gene regulation. J. Mol. Biol. 341:635, 2004. Mase, N., Thayumanavan, R., Tanaka, F., Barbas, C.F. III. Direct asymmetric organocatalytic Michael reactions of α,α-disubstituted aldehydes with β-nitrostyrenes for the synthesis of quaternary carbon-containing products. Org. Lett. 6:2527, 2004. Notz, W., Tanaka, F., Barbas, C.F. III. Enamine-based organocatalysis with proline and diamines: the development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels-Alder reactions. Acc. Chem. Res. 37:580, 2004. Notz, W., Watanabe, S., Chowdari, N.S., Zhong, G., Betancort, J.M., Tanaka, F., Barbas, C.F. III. The scope of the direct proline-catalyzed asymmetric addition of ketones to imines. Adv. Synth. Catal. 346:1131, 2004. Popkov, M., Jendreyko, N., McGavern, D.B., Rader, C., Barbas, C.F. III. Targeting tumor angiogenesis with adenovirus-delivered anti-Tie-2 intrabody. Cancer Res. 65:972, 2005. Popkov, M., Rader, C., Barbas, C.F. III. Isolation of human prostate cancer cell reactive antibodies using phage display technology. J. Immunol. Methods 291:137, 2004. Ramachary, D.B., Barbas, C.F. III. Direct amino acid-catalyzed asymmetric desymmetrization of meso-compounds: tandem aminoxylation/O-N bond heterolysis reactions. Org. Lett. 7:1577, 2005. Ramachary, D.B., Barbas, C.F. III. Towards organo-click chemistry: development of organocatalytic multicomponent reactions through combinations of aldol, Wittig, Knoevenagel, Michael, Diels-Alder and Huisgen cycloaddition reactions. Chemistry 10:5323, 2004. Sinha, S.C., Li, l.-S., Watanabe, S., Kaltgrad, E., Tanaka, F., Rader, C., Lerner, R.A., Barbas, C.F. III. Aldolase antibody activation of prodrugs of potent aldehydecontaining cytotoxics for selective chemotherapy. Chemistry 10:5467, 2004. Steiner, D.D., Mase, N., Barbas, C.F. III. Direct asymmetric α-fluorination of aldehydes. Angew. Chem. Int. Ed. 44:3706, 2005. Suri, J.T., Ramachary, D.B., Barbas, C.F. III. Mimicking dihydroxy acetone phosphate-utilizing aldolases through organocatalysis: a facile route to carbohydrates and aminosugars. Org. Lett. 7:1383, 2005. Tanaka, F., Barbas, C.F. III. Enamine-based reactions using organocatalysts: from aldolase antibodies to small amino acid and amine catalysts. J. Synth. Org. Chem. Jpn., in press. Tanaka, F., Barbas, C.F. III. Organocatalytic approaches to enantioenriched β-amino acids. In: Enantioselective Synthesis of β-Amino Acids, 2nd ed. Juaristi, E., Soloshonok, V. (Eds.). Wiley-VCH, New York, 2004, p. 195. Tanaka, F., Barbas, C.F. III. Reactive immunization: a unique approach to aldolase antibodies. In: Catalytic Antibodies. Keinan, E. (Ed.). Wiley-VCH, New York, 2004, p. 304. Tanaka, F., Flores, F., Kubitz, D., Lerner, R.A., Barbas, C.F. III. Antibody-catalyzed aminolysis of a chloropyrimidine derivative. Chem. Commun. (Camb.) 1242, 2004, Issue 10. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Synthetic Enzymes, Catalytic Antibodies, Ozone Scavengers, Organic Synthesis, and Biomolecular Computing E. Keinan, C.H. Lo, H. Han, S. Sasmal, S. Ledoux, N. Metanis, G. Sklute, E. Kossoy, M. Soreni, D. Vebenov, R. Piran, M. Sinha, A. Alt, I. Ben-Shir, R. Girshfeld, S. Yogev e focus on synthetically modified enzymes, antibody-catalyzed reactions, anticancer and antiasthma agents, and biomolecular computation, as illustrated in the following examples. W SYNTHETIC ENZYMES Efforts to generate new enzymatic activities from existing protein scaffolds may not only provide biotechnologically useful catalysts but also lead to better understanding of the natural process of evolution. We profoundly changed the catalytic activity and mechanism of the enzyme 4-oxalocrotonate tautomerase by means of rationally designed synthetic mutations. For example, a single amino acid substitution that corresponds to a mutation in a single base pair led to a dramatic change in the catalytic activity. Although the wild-type enzyme catalyzes only the tautomerization of 4-oxalocrotonate, the mutant P1A catalyzes both the original tautomerization reaction via a general acid-base mechanism and the decarboxylation of oxaloacetate via a nucleophilic mechanism. The observation that a single catalytic group in an enzyme can catalyze 2 reactions by 2 different mechanisms supports the hypothesis that enzyme evolution is a continuum in which a new catalytic mechanism is gained while the parent activity declines gradually through small changes in the amino acid sequence of the primordial enzyme. 214 MOLECULAR BIOLOGY 2005 We also showed that the electrostatic manipulation of an enzyme’s active site can alter the substrate specificity of the enzyme in a predictable way. We replaced 1, 2, or all 3 active-site arginine residues with citrulline analogs to maintain the steric features of the active site of 4-oxalocrotonate tautomerase while changing its electronic properties. These synthetic changes revealed that the wild-type enzyme binds the natural substrate predominantly through electrostatic interactions. This and other mechanistic insights led to the design of a modified enzyme that was specific for a new substrate that had different electrostatic properties and that bound the enzyme via hydrogen-bonding complementarity rather than electrostatic interactions. The synthetic analog of the natural 4-oxalocrotonate tautomerase was a poor catalyst of the natural 4-oxalocrotonate substrate but an efficient catalyst for a ketoamide substrate. This research on synthetic enzymes is being done in collaboration with P.E. Dawson, Department of Cell Biology. C ATA LY T I C A N T I B O D I E S Although the solution photochemical reaction of the ketone 1 (in Fig. 1) yields only the cleavage products 2 and 3, in the presence of 20F10, an antibody to 5a and 5b, this Norrish type II reaction results in the selective formation of cis-cyclobutanol (compound 4 in Fig. 1). Furthermore, the fact that compound 4, which consists of 2 asymmetric centers, is obtained as a single diastereomer makes this photoproduct a valuable building block for the synthesis of natural products. Another reaction that is exclusively catalyzed by 20F10 is the photochemical formation of cyclopropanol products. An aldolase antibody, 24H6, obtained from immunization with large diketone haptens has an active-site lysine residue with a perturbed pKa of 7.0. This antibody catalyzes both the aldol addition and the retrograde aldol fragmentation with a broad range of substrates that differ structurally from the hapten. This observation suggests that in reactive immunization with 1,3-diketones, the hapten structure governs the chemistry but not the overall organization of the active site. Antibody 24H6 also catalyzes the oxidation of α-hydroxyketones to α-diketones. The deuterium exchange at the α position of many ketones and aldehydes is also efficiently catalyzed by aldolase antibodies 38C2 and 24H6. All reactions were carried out in deuterium oxide under neutral conditions and showed regioselectivity, chemoselectivity, and high catalytic rates. O Z O N E S C AV E N G E R S A N D A N T I A S T H M A A C T I V I T Y A new hypothesis we proposed for the mechanism of asthmatic inflammation has led to an ozone-scavenging compound that prevents bronchial obstruction in rats with asthma. Previously, scientists at Scripps Research discovered that ozone can be generated not only via the antibody-mediated water oxidation pathway but also by antibody-coated activated white blood cells during inflammatory processes. This finding led us to speculate that the pulmonary inflammation in asthma might be caused by ozone production by white blood cells in lungs and that inhalation of electron-rich olefins, which are known ozone scavengers, might have antiasthmatic effects. In experiments in rats, inhalation of such a compound, limonene, caused a significant improvement in asthmatic symptoms. These results could have consequences in the management of asthma. ORGANIC SYNTHESIS F i g . 1 . The photochemical Norrish type II reaction of ketone 1 produces in solution the cleavage products 2 and 3. Antibody 20F10, which was elicited against a mixture of 5a and 5b, catalyzes enantioselective formation of cis-cyclobutanol (4). Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Annonaceous acetogenins, particularly those with adjacent bis-tetrahydrofuran rings, have remarkable cytotoxic, antitumor, antimalarial, immunosuppressive, pesticidal, and antifeedant activities. More than 350 different acetogenins have been isolated from only 35 of 2300 plants of the family Annonaceae. We developed synthetic approaches that can be used to generate chemical libraries of stereoisomeric acetogenins. These efforts resulted in the total synthesis of several naturally occurring acetogenins, including asimicin, bullatacin, trilobacin, rolliniastatin, solamin, reticulatacin, rollidecins C and D, goniocin, cyclogoniodenin, and mucocin, and many nonnatural stereoisomers. A substituted photoactive derivative of asimicin has been MOLECULAR BIOLOGY 2005 prepared for photoaffinity labeling of the target protein subunit in the mitochondrial complex I. This research is being done in collaboration with S.C. Sinha, Department of Molecular Biology. 215 This technology allowed parallel computation and automatic, real-time detection with DNA chips that carry multiple input molecules and can be used as pixel arrays for image encryption. BIOMOLECULAR COMPUTING DEVICES Four years ago we described the first nanoscale, programmable finite automaton with 2 symbols and 2 states that computed autonomously. All of the components of the device, including hardware, software, input, and output, were biomolecules mixed together in solution. The hardware consisted of a restriction nuclease and a ligase; the software (transition rules) and the input were double-stranded DNA oligomers (Fig. 2). PUBLICATIONS Dubnikova, F., Kosloff, R., Almog, J., Zeiri, Y., Boese, R., Itzhaky, H., Alt, A., Keinan, E. Decomposition of triacetone triperoxide is an entropic explosion. J. Am. Chem. Soc. 127:1146, 2005. Keinan, E., Alt, A., Amir, G., Bentur, L., Bibi, H., Shoseyov, D. Natural ozone scavenger prevents asthma in sensitized rats. Bioorg. Med. Chem. 13:557, 2005. Metanis, N., Keinan, E., Dawson, P.E. A designed synthetic analogue of 4-OT is specific for a non-natural substrate. J. Am. Chem. Soc. 127:5862, 2005. Saphier, S., Hu, Y., Sinha, S.C., Houk, K.N., Keinan, E. The origin of selectivity in the antibody 20F10-catalyzed Yang cyclization. J. Am. Chem. Soc. 127:132, 2005. Soreni, M., Yogev, S., Kossoy, E., Shoham, Y., Keinan, E. Parallel biomolecular computation on surfaces with advanced finite automata. J. Am. Chem. Soc. 127:3935, 2005. Antibody Catalysis and Organic Synthesis S.C. Sinha, R.A. Lerner, S. Das, S. Abraham, F. Guo, Z. Chen ur main research interests are antibody catalysis and the applications of antibody catalysts in organic synthesis, prodrug activation, and the development of cell-targeting antibody constructs. In addition, we also focus on synthetic and medicinal chemistry, including the total synthesis of biologically important natural products and synthetic compounds and their analogs and new methods of synthesis. O F i g . 2 . A biomolecular computing machine made of molecules. The hardware consists of a restriction nuclease and a ligase; the input, transition molecules (software), and detection molecules are all made of double-stranded DNA. A N T I B O D Y C ATA LY S I S A N D I T S A P P L I C AT I O N S Computation was carried out by processing the input molecule via repetitive cycles of restriction, hybridization, and ligation reactions to produce a final-state output in the form of a double-stranded DNA molecule. Currently, we are taking the concept of molecular computing a step further and are constructing computing devices in which the computation output is a specific biological function rather than a specific molecule. Most recently, we markedly increased the levels of complexity and mathematical power of these automata by the design of a 3-state–3-symbol automaton, thus increasing the number of syntactically distinct programs from 765 to 1 billion. We have further amplified the applicability of this design by using surface-anchored input molecules and surface plasmon resonance technology to monitor the computation steps in real time. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Aldolase antibodies 38C2, 84G3, and 93F3 produced by the reactive immunization technique are highly useful in synthetic organic chemistry, as indicated by their application in the syntheses of a number of natural products, including epothilones. These antibodies catalyze both aldol and retro-aldol reactions and yield products with high enantioselectivities. The high catalytic rate of the retro-aldol reaction makes the antibodies useful in prodrug therapy. In prodrug therapy, an enzyme or a catalytic antibody is used to activate a nontoxic prodrug at a targeted site, thereby producing a cytotoxic drug. We are developing prodrugs of cytotoxic molecules, including paclitaxel, doxorubicin analogs, enediynes, CBI analogs, and epothilones, that can be activated efficiently by aldolase antibodies. In particular, we prepared and evaluated 216 MOLECULAR BIOLOGY 2005 several prodrugs of the analogs of dynemicin B and doxorubicin. The prodrugs of dynemicin analogs are activated by using antibody 38C2; those of doxorubicin analogs, by 93F3 (Fig. 1). On the basis of these studies, we are developing new linkers for the prodrugs so that activation of the prodrugs can be selectively achieved at high catalytic rate. F i g . 3 . Structure of sorangiolides A and B (top) and a general structure of bis-tetrahydrofuran annonaceous acetogenins (bottom). F i g . 1 . Structure of the prodrugs of doxorubicin (DOX) analogs. Using antibody 38C2, we also developed antagonist-38C2 conjugates. The conjugates bound efficiently to cells expressing the integrins αvβ3 and αvβ5. The conjugates have several advantages, including prolongation of half-life of the antagonist and in vivo assembly of the conjugate. On the basis of our initial studies, in collaboration with C.F. Barbas, Department of Molecular Biology, we synthesized a series of antagonist-38C2 conjugates and evaluated them by using breast cancer cell lines that express the integrins αvβ3 and αvβ5. Several conjugates (Fig. 2) bound to these cell lines with high affinity. Our findings, which were supported by molecular docking studies, provided preliminary information on how the compounds should be derivatized. active against gram-positive bacteria. Our goal is to synthesize analogs of sorangiolides that are highly active. The bis-tetrahydrofuran acetogenins are among the most active cancer agents and are toxic to a number of human cancer cell lines at much lower concentrations than doxorubicin is. In collaboration with E. Keinan, Department of Molecular Biology, we synthesized an analog of asimicin, an annonaceous acetogenin, for photoaffinity labeling of the corresponding receptor. In other studies, we developed a bidirectional approach for the synthesis of all 64 diastereomers of the adjacent bis-tetrahydrofuran acetogenins (Fig. 3). Starting with 8 diene lactones, we synthesized 36 bifunctional adjacent bis-tetrahydrofuran lactones by using 5 key reactions: (1) monooxidative or bis-oxidative cyclization mediated by rhenium(VII) oxides, (2) Shi monoasymmetric or bis-asymmetric epoxidation, (3) Sharpless asymmetric dihydroxylation, (4) Williamson-type etherification, and (5) Mitsunobu inversion. Further studies are in progress. PUBLICATIONS Li, L.-S., Rader, C., Matsushita, M., Das, S., Barbas, C.F. III, Lerner, R.A., Sinha, S.C. Chemical-adaptor immunotherapy: design, synthesis and evaluation of novel integrin-targeting devices. J. Med. Chem. 47:5630, 2004. Saphier, S., Hu, Y., Sinha, S.C., Houk, K.N., Keinan, E. Origin of selectivity in the antibody 20F10-catalyzed Yang cyclization. J. Am. Chem. Soc. 127:132, 2005. Sinha, S.C., Li, L.-S., Watanabe, S.-I., Kaltgrad, E., Tanaka, F., Rader, C., Lerner, R.A., Barbas, C.F. III. Aldolase antibody activation of prodrugs of potent aldehydecontaining cytotoxics for selective chemotherapy. Chemistry 10:5467, 2004. F i g . 2 . Structure of the compounds that target the integrins αvβ3 and αvβ5 for conjugation with 38C2. S Y N T H E S I S O F N AT U R A L P R O D U C T S A N D T H E I R ANALOGS In the past year, we focused on the synthesis of naturally occurring macrocyclic molecules, sorangiolides A and B, and the library of bis-tetrahydrofuran annonaceous acetogenins. Sorangiolides (Fig. 3) are weakly Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. MOLECULAR BIOLOGY 2005 Structure, Function, and Applications of Virus Particles J.E. Johnson, L. Basumallic, A. Chatterji, W. FernandezOchoa, L. Gan, I. Gertsman, R. Khayat, J. Lanman, K. Lee, T. Matsui, P. Natarajan, A. Odegard, J. Speir, L. Tang, H. Walukiewicz, E. Wu e investigate model virus systems that provide insights for understanding assembly, maturation, entry, localization, and replication of nonenveloped viruses. We also have developed viruses as reagents for applications in nanotechnology, chemistry, and biology. We investigate viruses that infect bacteria, insects, yeast, plants, and, recently, the extreme thermophile Sulfolobus. These viruses have genomes of single-stranded RNA, double-stranded RNA, and double-stranded DNA. In many instances, we use viruslike particles that do not contain infectious genomes. We use a variety of physical methods to investigate structure-function relationships, including single-crystal and static and time-resolved solution x-ray diffraction, electron cryomicroscopy and image reconstruction, mass spectrometry, structure-based computational analyses, and methods associated with thermodynamic characterization of virus particles and their transitions. Biological methods we use include genetic engineering of viral genes and their expression in Escherichia coli, mammalian cells, insect cells, and yeast and the characterization of these gene products by the physical methods mentioned previously. For cytologic studies of viral entry and infection, we use fluorescence and electron microscopy and particles assembled in heterologous expression systems. Our studies depend on extensive consultations and collaborations with others at Scripps Research, including groups led by C.L. Brooks, D.A. Case, B. Carragher, M.G. Finn, M.R. Ghadiri, T. Lin, M. Manchester, D.R. Millar, R.A. Milligan, C. Potter, V. Reddy, A. Schneemann, G. Siuzdak, K.F. Sullivan, J.R. Williamson, and M.J. Yeager, and a variety of groups outside of Scripps. W DOUBLE-STRANDED DNA VIRUSES HK97 is a double-stranded DNA bacterial virus similar to phage λ. It undergoes a remarkable morphogenesis in its assembly and maturation, and this process can be recapitulated in vitro. We determined the atomic resolution structure of the 650-Å mature head II particle and discovered the mechanism used to concatenate the subunits of the particle into a chain-mail Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 217 fabric similar to that seen in the armor of medieval knights. We created a model of the procapsid on the basis of the 5-Å electron cryomicroscopy structure in which the coordinates from the head II particle were readily fitted. Recently, we used single-value decomposition analysis of time-resolved solution x-ray scattering data and single-molecule fluorescence to show that the initial maturation of prohead II (~470 Å in diameter) to expansion intermediate I (546 Å in diameter) occurs as a highly cooperative, stochastic event with no significantly populated intermediates and takes less than 1 second for an individual particle. Bacteriophage P22 is the prototype of the Podoviridae, which are characterized by a T = 7 capsid with a short tail structure incorporated into a unique 5-fold vertex. We previously determined the icosahedrally averaged structure of the capsid at 20-Å resolution, the 10-Å structure of the connector protein, and the 20-Å structure of the tail machine. Recently, we did a reconstruction of the virus without imposing symmetry, enabling us to visualize the detailed relationship of all these components (Fig. 1). F i g . 1 . Electron cryomicroscopy reconstruction of the bacterio- phage P22. The reconstruction was done with 1800 particles and no applications of symmetry. The reconstructed density required first generating an icosahedrally averaged electron density that ignored the tail assembly and then inserting the tail assembly (determined as a separate reconstruction) into the icosahedral density to create a tailed phage model. The model was then back projected in all icosahedral orientations onto each individual particle, and the orientation that gave the highest correlation coefficient (i.e., aligned the tails) was used to reconstruct the final density. Note that without any application of symmetry, both the tail machine and the T = 7 surface lattice are clearly defined in the reconstructed density. 218 MOLECULAR BIOLOGY 2005 Sulfolobus turreted icosahedral virus is an archaeal virus isolated from Sulfolobus, which grows in the acidic hot sulfur springs (pH 2–4, 72°C–92°C) in Yellowstone National Park. An electron cryomicroscopy reconstruction of the virus showed that the capsid has pseudo T = 31 quasi symmetry and is 1000 Å in diameter, including the pentons. We solved the x-ray structure of the major capsid protein of the virus and revealed a fold nearly identical to the major capsid proteins of the eukaryotic adenoviruses; PBCV-1, a virus that infects fresh water algae; and PRD-1, a virus that infects bacteria. These findings indicate a virus phylogeny that spans the 3 domains of life (Eucarya, Bacteria, and Archaea) and suggests that these viruses may be related to a virus that preceded the division of life into 3 domains more than 3 billion years ago. Blum, A.S., Soto, C.M., Wilson, C.D., Cole, J.D., Kim, M., Gnade, B., Chatterji, A., Ochoa, W.F., Lin, T., Johnson, J., Ratna, B.R. Cowpea mosaic virus as a scaffold for 3-D patterning of gold nanoparticles. Nano Lett. 4:867, 2004. Bothner, B., Taylor, D., Jun, B., Lee, K.K., Siuzdak, G., Schultz, C.P., Johnson, J.E. Maturation of a tetravirus capsid alters the dynamic properties and creates a metastable complex. Virology 334:17, 2005. Chatterji, A., Ochoa, W.F., Paine, M., Ratna, B.R., Johnson, J.E., Lin, T. New addresses on an addressable virus nanoblock; uniquely reactive Lys residues on cowpea mosaic virus. Chem. Biol. 11:855, 2004. Chatterji, A., Ochoa, W.F., Ueno, T., Lin, T., Johnson, J.E. A virus-based nanoblock with tunable electrostatic properties. Nano Lett. 5:597, 2005. Falkner, J.C., Turner, M.E., Bosworth, J.K., Trentler, T.J., Johnson, J.E., Lin, T., Colvin, V.L. Virus crystals as nanocomposite scaffolds. J. Am. Chem. Soc. 127:5274, 2005. Girard, E., Kahn, R., Mezouar, M., Dhaussy, A.C., Lin, T., Johnson, J.E., Fourme, R. The first crystal structure of a complex macromolecular assembly under high pressure: CpMV at 330 MPa. Biophys. J. 88:3562, 2005. Johnson, K.N., Tang, L., Johnson, J.E., Ball, L.A. Heterologous RNA encapsidated in Pariacoto virus-like particles forms a dodecahedral cage similar to genomic RNA in wild-type virions. J. Virol. 78:11371, 2004. SINGLE-STRANDED RNA VIRUSES Flock House virus is a single-stranded RNA virus that infects Drosophila. We are studying viral entry and early expression and assembly of the capsid protein. Recently, studies on viral entry indicated the presence of an “eluted” particle early in infection that has initiated its disassembly program but is then eluted back into the medium. We did a phenotypic characterization of the particles, and we are using electron cryomicroscopy to study them. For studies on the expression and assembly of the capsid protein, we are using tags genetically inserted in the capsid protein that allow the freshly made proteins to be optically visualized with a fluorophore and in the electron microscope with photoconversion of the fluorophore. Tetraviruses are single-stranded RNA viruses that infect Lepidoptera. Expression of the capsid protein in the baculovirus system leads to spontaneous assembly of viruslike particles that we can investigate in vitro. The particles exist as procapsids (480 Å) at pH 7 and as capsids (410 Å) at pH 5. We used limited proteolysis and mass spectrometry to investigate the driving force of the transition, the mechanism of an autocatalytic cleavage, and the dynamic features of both forms. Cowpea mosaic virus is a 30-nM reagent that we use for chemistry and nanotechnology. In collaboration with T. Lin, Department of Molecular Biology, we generated and produced a large variety of viable mutations of the virus in gram quantities for nanopatterning, molecular electronic scaffolds, and platforms for novel chemistry. PUBLICATIONS Blum, A.S., Soto, C.M. Wilson, C.D., Brower, T.L., Pollack, S.K., Schull, T.L., Chatterji, A., Lin, T., Johnson, J.E., Amsinck, C., Franzon, P., Shashidhar, R., Ratna, B.R. An engineered virus as a scaffold for three-dimensional self-assembly on the nanoscale. Small 1:702, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Lin, T., Lomonossoff, G.P., Johnson, J.E. Structure-based engineering of an icosahedral virus for nanomedicine and nanotechnology. In: Nanotechnology in Biology and Medicine: Methods, Devices, and Applications. Vo-Dinh, T. (Ed.). CRC Press, Boca Raton, FL, in press. Lin, T., Schildkamp, W., Brister, K., Doerschuk, P.C., Somayazulu, M., Mao, H.K., Johnson, J.E. The mechanism of high pressure induced ordering in a macromolecular crystal. Acta Crystallogr. D Biol. Crystallogr. 61:737, 2005. Medintz, I., Mattoussi, H., Sapsford, K., Chatterji, A., Johnson, J.E. Decoration of discretely immobilized cowpea mosaic virus with luminescent quantum dots. Langmuir, in press. Natarajan, P., Lander, G., Shepherd, C., Reddy, V., Brooks, C.L. III, Johnson, J.E. Virus Particle Explorer (VIPER), a Web-based repository of virus structural data and derived information. Nat. Microbiol. Rev., in press. Reddy, V., Schneemann, A., Johnson, J.E. Nodavirus endopeptidase. In: Handbook of Proteolytic Enzymes, 2nd ed. Barret, A.J., Rawlings, N.D., Woessner, J.F. (Eds.). Academic Press, San Diego, 2004, Vol. 2, p. 198. Reddy, V.S., Natarajan, P., Lander, G., Qu, C., Brooks, C.L. III, Johnson, J.E. Virus Particle Explorer (VIPER): a repository of virus capsid structures. In: Conformational Proteomics of Macromolecular Architecture: Approaching the Structure of Large Molecular Assemblies and Their Mechanisms of Action. Cheng, R.H., Hammar, L. (Eds.). World Scientific, River Edge, NJ, 2004, p. 403. Schwarcz, W.D., Barroso, S.P., Gomes, A.M., Johnson, J.E., Schneemann, A., Oliveira, A.C., Silva, J.L. Virus stability and protein-nucleic acid interaction as studied by high-pressure effects on nodaviruses. Cell. Mol. Biol. (Noisy-le-grand) 50:419, 2004. Strable, E., Johnson, J.E., Finn, M.G. Natural supramolecular building blocks: icosahedral virus particles organized by attached oligonucleotides. Nano Lett. 4:1385, 2004. Tang, J., Naitow, H., Gardner, N.A., Kolesar, A., Tang, L., Wickner, R.B., Johnson, J.E. The structural basis of recognition and removal of cellular mRNA 7-methyl G “caps” by a viral capsid protein: a unique viral response to host defense. J. Mol. Recognit. 18:158, 2005. Tang, L., Marion, W.R., Cingolani, G., Prevelige, P.E., Johnson, J.E. The three-dimensional structure of the bacteriophage P22 tail machine. EMBO J. 24:2087, 2005. Taylor, D.J., Johnson, J.E. Folding and particle assembly are disrupted by singlepoint mutations near the autocatalytic cleavage site of nudaurelia capensis 4 virus capsid protein. Protein Sci. 14:401, 2005. Taylor, D.J., Speir, J., Reddy, V., Cingolani, G., Pringle, F., Ball, L.A., Johnson, J.E. Preliminary x-ray characterization of authentic providence virus and attempts to express its coat protein gene in recombinant baculovirus. Arch. Virol., in press. MOLECULAR BIOLOGY 2005 An Icosahedral Scaffold for Biophysical Studies and Nanomanufacturing T. Lin, J.E. Johnson, A. Chatterji, W.F. Ochoa, A. Stone, T. Ueno owpea mosaic virus (CPMV) is an icosahedral plant virus with a diameter of 30 nm. Because of its exceptional stability, high yield, ease of production, structural information to the level of atomic definition, and accessible genetic programmability, the virus has been used as a model system for biophysical studies and has been engineered for applications in biotechnology and nanotechnology. C A S S E M B LY O F N A N O M AT E R I A L S O N A N ICOSAHEDRAL SCAFFOLD A quintessential tenet of nanotechnology is the selfassembly of components at nanometer scale to form devices. Although small molecules with novel electronic properties can be synthesized, it is generally difficult to get functional connectivity among the different components in designed patterns. In contrast, because of their versatility, programmability through genetic engineering, and propensity to form arrays, biological macromolecules are more amenable for self-assembly either as devices for direct use or as scaffolds for patterning small molecules. We showed that CPMV can be used as a template for nanochemistry by introducing unique cysteine residues and exploiting the native lysine residues. In collaboration with B.R. Ratna, Naval Research Laboratory, Washington, D.C., we used the viral capsid as a nano circuit board and the reactive groups as anchoring points for the assembly of electronic molecules, oligophenylene-vinylene and others. The establishment of the molecular network was demonstrated by measuring electronic conductance via scanning tunnel microscopy. 219 sure. The crystals assigned to the I23 space group diffracted x-rays to higher resolution than did those assigned to the P23 space group. The assignment of the P23 space group was due to the presence of reflections with indices h + k + l = (2n + 1) (odd reflections), which are forbidden in the I23 space group. Analysis of the odd reflections from the P23 crystals at atmospheric pressure indicated that they originated from a rotational disorder in the the I23 crystals. The odd reflections were eliminated by applying 3.5 kbar of pressure, which transformed the crystals from the apparently primitive cell to the body-centered I23 cell, with dramatic improvement in diffraction. PUBLICATIONS Blum, S.A., Soto, C.M., Wilson, C.D., Brower, T.L., Pollack, S.K., Schull, T.L., Chatterji, A., Lin, T., Johnson, J.E., Amsinck, C., Franson, P., Shashidhar, R., Ratna, B.R. An engineered virus as a scaffold for three-dimensional self-assembly on the nanoscale. Small 1:702, 2005. Chatterji, A., Ochoa, W., Shamieh, L., Salakian, S.P., Wong, S.M., Clinton, G., Ghosh, P., Lin, T., Johnson, J.E. Chemical conjugation of heterologous proteins on the surface of cowpea mosaic virus. Bioconjug. Chem. 15:807, 2004. Chatterji, A., Ochoa, W.F., Paine, M., Ratna, B.R., Johnson, J.E., Lin, T. New addresses on an addressable virus nanoblock: uniquely reactive Lys residues on cowpea mosaic virus. Chem. Biol. 11:855, 2004. Chatterji, A., Ochoa, W.F., Ueno, T., Lin, T., Johnson, J.E. A virus-based nanoblock with tunable electrostatic properties. Nano Lett. 5:597, 2005. Falkner, J.C., Turner, M.E., Bosworth, J.K., Trentler, T.J., Johnson, J.E., Lin, T., Colvin, V.L. Virus crystals as nanocomposite scaffolds. J. Am. Chem. Soc. 127:5274, 2005. Girard, E., Kahn, R., Mezouar, M., Dhaussy, A.-C., Lin, T., Johnson J.E., Fourme, R. The first crystal structure of a complex macromolecular assembly under high pressure: CpMV at 330 MPa. Biophys. J. 88:3562, 2005. Lin, T., Schildkamp, W., Brister, K., Doerschuk, P.C., Somayazulu, M., Mao H., Johnson, J.E. The mechanism of high-pressure-induced ordering in a macromolecular crystal. Acta Crystallogr. D Biol. Crystallogr. 61:737, 2005. Design and Informatics in Structural Virology V.S. Reddy, C.M. Shepherd, C. Hsu, S. Kumar, R. Mannige, I. Borelli, C.L. Brooks III, J.E. Johnson, M. Manchester, H I G H - P R E S S U R E C R Y S TA L L O G R A P H Y Using high pressure, we markedly improved the diffraction from the cubic crystals of CPMV from about 4-Å to 2.1-Å resolution. If this use of pressure is generally applicable, it can have a marked effect on structural biology. To this end, we carried out mechanistic studies of the pressure-induced rectification of crystal imperfection. Two types of cubic crystals were assigned to either an I23 or a P23 space group. The 2 types had the same rhombic dodecahedral morphology at atmospheric presPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. A. Schneemann e are interested in understanding the structural underpinnings and requirements for formation of viral capsids and in designing novel protein shells that polyvalently display molecules of interest. To this end, we use structural, computational, informatics, and genetic methods. Viruses are highly evolved macromolecular machines that perform a variety of functions during their life cycle, W 220 MOLECULAR BIOLOGY 2005 including selective packaging of the genome, selfassembly, binding to host cells, and delivery of the genome to the targeted cells. Simple viruses, such as nonenveloped viruses, form closed protein shells or capsids of uniform size and character by the self-association of structural and functional components: proteins and the nucleic acid genome. Hence, these viruses are useful for structural and functional analyses. To understand the requirements for formation of the closed protein shell in viral capsids in terms of structure and interactions, we established a repository of structurally characterized viral capsids in a relational database format, namely the Viper Particle Explorer (http://viperdb.scripps.edu). At the database, we use computational methods to analyze these protein shells in terms of protein-protein interactions: contacting residue pairs, association energies, individual residue contributions, and surface characteristics. To facilitate these studies, we are developing structural tools for analysis of viral structures as part of the Multiscale Modeling Tools for Structural Biology, the National Institutes of Health research resource headed by C.L. Brooks, Department of Molecular Biology. The structural and taxonomic data and the derived results are stored in a MySQL database for ease of querying and comparing the properties of interest within and across families of viruses. Furthermore, using the structural similarity that occurs within a virus family, we are building homology models for the uncharacterized members of virus families. These models will be useful for molecular virologists investigating structural and functional relationships in viruses. To generate novel reagents, such as vaccines and antitoxins against cytotoxins such as ricin and pathogens in general, we are expressing decoys of pathogenic molecules on the surfaces of viral capsids. Currently, tomato bushy stunt virus–like capsids are the display platform of choice; the platform consists of multiple copies of a 2-domain capsid protein subunit with the C-terminal P-domain exposed on the surface. Such a unique subunit structure is useful for attaching peptides or proteins of interest at the end of the C terminus of the capsid protein or for replacing the P-domain with the proteins of interest rather than inserting them in a loop. PUBLICATIONS Reddy, V.S., Johnson J.E. Structure-derived insights into virus assembly. Adv. Virus Res. 64C:45, 2005. Reddy, V.S., Schneemann, A., Johnson, J.E. Nodavirus endopeptidase. In: Handbook of Proteolytic Enzymes, 2nd ed. Barrett, A., Rawlings, N.D., Woessner, J.F. (Eds.). Academic Press, San Diego, 2004, p. 197. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Shepherd, C.M., Reddy, V.S. Extent of protein-protein interactions and quasi-equivalence in viral capsids. Proteins 58:472, 2005. Biology and Applications of Capsids of Icosahedral Viruses A. Schneemann, B. Groschel, J. Lee, D. Manayani, M. Siladi, P.A. Venter oat proteins of nonenveloped, icosahedral viruses perform multiple functions during the course of viral infection, including capsid assembly, specific encapsidation of the viral genome, binding to a cellular receptor, and uncoating. In some viruses, a single type of protein is sufficient to carry out these functions; we are interested in the determinants that endow a polypeptide chain with such versatility. We seek to harness this versatility for novel applications of viruses in biotechnology and nanotechnology. We focus on a structurally and genetically wellcharacterized virus family, the T = 3 nodaviruses. Nodaviruses are composed of 180 copies of a single coat protein and 2 strands of positive-sense RNA. Currently, we are elucidating the mechanism by which the 2 genomic RNAs are packaged into a single virion. Our long-term goal is to develop nodaviruses as RNA packaging and delivery vectors. Our data indicate that the 2 viral RNAs are recognized separately, but it is not yet known whether packaging occurs sequentially and whether one or more coat protein subunits are involved in this process. Interestingly, we recently discovered that packaging of the RNA genome is directly coupled to replication of the genome, suggesting potential approaches for packaging of foreign RNAs. In other studies, we are investigating the mechanism by which nodaviral protein B2 suppresses RNA silencing in infected cells. Preliminary data indicate that protein B2 binds to double-stranded RNA and that it interferes with cleavage of double-stranded RNA substrates by the cellular protein Dicer. We are also collaborating with several investigators at Scripps Research, the Salk Institute, and Harvard University to develop nodaviruses as platforms for delivery of anthrax antitoxins. To this end, we are using particles to display the VWA domain of capillary morphogenesis protein 2, the cellular receptor for anthrax toxin, in a multivalent fashion on the surface of the virion. Two insertion sites yielding different patterns of C MOLECULAR BIOLOGY 2005 180 copies of the VWA domain were selected on the basis of computational modeling of the high-resolution crystal structure of the insect nodavirus Flock House virus. The resulting chimeric viruslike particles protect cultured cells from the toxic effects of protective antigen and lethal factor, 2 of the 3 proteins that make up anthrax toxin. Experiments in animals are currently under way to show that these particles also function as antitoxins in vivo. This research is important because it illustrates that protein domains containing more than 150 amino acids can be displayed on Flock House virus in a biologically functional form, suggesting numerous additional applications. Flock House virus particles are also good candidates for novel materials in nanotechnology applications. The particles are stable, easily manipulated, biocompatible, and nontoxic in vivo and can be produced easily and in high quantities. The high-resolution x-ray structure of the virus revealed the potential for using chemical approaches to attach ligands to the surface of the virus and for using genetic strategies to modify the capsid. In collaboration with M. Manchester, Department of Cell biology, and M. Ozkan, University of California, Riverside, California, we used conjugation chemistry to couple inorganic nanotubes and quantum dots to Flock House virus particles to produce an array of novel hybrid structures. This approach may one day be used to fabricate unique materials for a variety of applications, including biofilms with tunable pore sizes, 3-dimensional scaffolds for production of nanoelectronic devices, and drug delivery. PUBLICATIONS Portney, N.G., Singh, K., Chaudhary, S., Destito, G., Schneemann, A., Manchester, M., Ozka, M. Organic and inorganic nanoparticle hybrids. Langmuir 21:2098, 2005. Schwarcz, W.D., Barroso, S.P., Gomes, S.M., Johnson, J.E., Schneemann, A., Oliveira, A.C., Silva, J.L. Virus stability and protein-nucleic acid interaction as studied by high-pressure effects on nodaviruses. Cell. Mol. Biol. (Noisy-le-grand). 50:419, 2004. Venter, P.A., Krishna, N.K., Schneemann, A. Capsid protein synthesis from replicating RNA directs specific packaging of the genome of a multipartite, positivestrand RNA virus. J. Virol. 79:6239, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 221 Molecular Biology of Retroviruses J.H. Elder, A.P. de Parseval, Y.-C. Lin, S. de Rozieres, U. Chatterji,* K. Tam, B.E. Torbett** * Department of Immunology, Scripps Research ** Department of Molecular and Experimental Medicine, Scripps Research ur research centers on the molecular characterization of retroviruses, with emphasis on feline immunodeficiency virus (FIV). FIV causes an AIDS-like syndrome in domestic cats, and although it does not infect humans, the feline retrovirus has many structural and functional similarities to HIV, the causative agent of AIDS in humans. Thus, study of FIV can yield insights into ways to interfere with the retrovirus life cycle that may ultimately result in the development of treatments for infections in both cats and humans. During the past year, we focused on 2 major areas: the molecular characterization of cell-surface receptors for FIV and the molecular basis for the development of drug resistance in the aspartic protease encoded by FIV. O RECEPTOR STUDIES Like many strains of HIV, FIV uses the chemokine receptor CXCR4 to enter the primary target cell, the CD4 + T cell. However, unlike HIV, FIV does not use the cell-surface protein CD4 as a primary binding receptor. Rather, the feline lentivirus initially binds to another cell-surface molecule, CD134. In the past year, we characterized the expression of CD134 and showed that it is upregulated on CD4+ T cells. This observation explains why FIV can infect and kill this subset of T cells even though the virus’s surface glycoprotein does not interact with CD4. In an extension of these studies, we found that interaction of the FIV surface glycoprotein gp95 with a soluble version of CD134 allows the productive infection of cells that bear the entry receptor, CXCR4, but lack surface expression of the binding receptor, CD134. The results are consistent with the notion that binding of CD134 causes a conformational change in gp95, which in turn increases the affinity of interaction with CXCR4 and facilitates infection of the target cell. These effects are similar to the effects of binding of soluble CD4 by gp120, the cell-surface glycoprotein of HIV, and indicate that although different molecules are involved, the actual mechanisms of infection of FIV and HIV are strikingly similar. We speculate that the benefit of this type of binding cascade is to limit exposure of critical 222 MOLECULAR BIOLOGY 2005 regions of the surface glycoproteins to the immune system until the primary binding event has already occurred, thus reducing the likelihood of virus neutralization. We also precisely mapped regions of CD134 involved in interaction with gp95. CD134 is a member of the TNF-α receptor superfamily and has a domain structure similar to that of the TNF-α receptor. Human CD134 does not bind FIV gp95, even though human CD134 shares considerable amino acid homology with feline CD134. Using chimeric proteins consisting of feline and human CD134 and site-directed mutagenesis, we showed that as few as 3 amino acids in the C-terminal part of outer domain 1 of feline CD134 are sufficient to impart FIV gp95 binding and receptor function to human CD134. Structural studies of both receptor and ligand will establish a molecular basis for the putative conformational change induced by interaction with the binding receptor. D E V E L O P M E N T O F D R U G R E S I S TA N C E B Y F I V ASPAR TIC PROTEASE The aspartic protease of lentiviruses is a particularly good target for drug therapy because its function in processing the viral Gag and Pol polyproteins is absolutely required for generation of infectious virus. Drugs active against the HIV protease have been keys to the success of highly active antiretroviral therapy used to treat patients infected with HIV. The substrate and inhibitor specificity of FIV differs from that of HIV, and we previously reported the identification of amino acids that define the different specificities. Comparing FIV with HIV offers a means to better understand the development of resistance to therapy, an ongoing problem with current drugs used to treat HIV disease. Interestingly, parallels exist between amino acid positions that dictate differences in substrate specificity between FIV and HIV aspartic protease and those that mutate in response to drug treatment. Mutations in these sites increase the dissociation constant for complexes consisting of the protease and an inhibitor drug, but at a cost in catalytic efficiency for the protease. Over time, compensatory amino acid substitutions occur that result in an increase in catalytic efficiency, which results in increased expression of virus despite drug treatment. We prepared mutants of FIV protease in which amino acids found in drug-resistant HIV protease were placed in the equivalent positions in the FIV enzyme. These “HIV-inized” FIV proteases had drug sensitivity profiles similar to those of HIV protease. In studies with cells Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. transduced with gag/pol gene expression vectors encoding HIV-FIV hybrid proteases, the Gag/Pol polyproteins were processed with proper fidelity and had the expected drug sensitivities. When engineered into FIV, these hybrid proteases will offer a means to study drug resistance and to develop new inhibitors capable of blocking replication of drug-resistant viruses, without the biohazard associated with handling infectious HIV. PUBLICATIONS de Parseval, A., Chatterji, U., Morris, G., Sun, P., Olson, A.J., Elder, J.H. Structural mapping of CD134 residues critical for interaction with feline immunodeficiency virus. Nat. Struct. Mol. Biol. 12:60, 2005. de Parseval, A., Chatterji, U., Sun, P., Elder, J.H. Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc. Natl. Acad. Sci. U. S. A. 101:13044, 2004. de Rozieres, S., Swan, C.H., Sheeter, D.A., Clingerman, K.J., Lin, Y.-C., HuitrónReséndiz, S. Henriksen, S., Torbett, B.E., Elder, J.H. Assessment of FIV-C infection of cats as a function of treatment with the protease inhibitor, TL-3. Retrovirology 1:38, 2004. Montes-Rodriguez, C.J., Alavez, S., Elder, J.H., Haro, R., Moran, J., ProsperoGarcia, O. Prolonged waking reduces human immunodeficiency virus glycoprotein 120- or tumor necrosis factor α-induced apoptosis in the cerebral cortex of rats. Neurosci. Lett. 360:133, 2004. Metalloenzyme Engineering D.B. Goodin, C.D. Stout, A.-M.A. Hays, S. Vetter, E.C. Glazer, A.E. Pond, H.B. Gray,* J.R. Winkler,* J.H. Dawson,** T.L. Poulos,*** M.A. Marletta**** * California Institute of Technology, Pasadena, California ** University of South Carolina, Columbia, South Carolina *** University of California, Irvine, California **** University of California, Berkeley, California ur overall goals are to understand the fundamental structural features of metalloenzyme catalysts and to create catalysts for useful chemical reactions. We use a number of techniques in structural biology and spectroscopy and strategies of rational protein redesign and molecular evolution. In the past year, we made progress in several areas. One area of recent interest has been the design and use of molecular wires as probes for the active sites of enzymes such as cytochrome P450 and nitric oxide synthase (NOS). In an ongoing collaboration with H.B. Gray, California Institute of Technology, we are investigating the binding of these wires, which are specifically designed substrate analogs linked to photochemical or redox-active sensitizers, to the active site of metalloproteins. The wires are being developed to serve as reporters of the active-site environment and O MOLECULAR BIOLOGY 2005 as tools to allow rapid deposition or withdrawal of electrons to drive redox catalysis. In addition, luminescent wires that are quenched upon either binding or release from the protein may be useful as imaging agents or as tools for identifying novel enzyme inhibitors. P450s make up a large family of enzymes responsible for a vast range of biologically important oxidation reactions in mammals, plants, fungi, and bacteria. An important unresolved question concerns how the deeply buried heme cofactor of these enzymes achieves regioselective and stereoselective catalysis of a wide range of substrates. In the past year, we completed a detailed structural analysis by x-ray crystallography of cytochrome P450cam complexed with 2 sensitizer-linked substrate probes, D4A and D8A. These probes differ in length but bind identically at the substrate end of the wire (Fig. 1). 223 entry and product egress. The conformational change associated with movement of the F and G helices is transmitted to a backbone carbonyl at the active site of the enzyme, which has been implicated in gating the critical peroxy-bond cleavage that activates the enzyme for catalysis. In other studies, we are designing and synthesizing specific pterin-based molecular wires for the active site of NOS. NOSs are complex enzymes used for the production of nitric oxide from arginine and play many critical roles in biological signal transduction. As thiolate coordinate heme enzymes, they have structural and functional similarities to P450s. One unique feature is the role played by the pterin cofactor of NOS. Recent results suggest that the pterin donates an electron to either the heme or the substrate at defined steps in the catalytic mechanism. In the past year, we designed and synthesized a series of pterin analogs tethered to sensitizers containing redox-active ruthenium to be used as specific molecular triggers and probes of the NOS active site. In addition, we measured the FeIII/II and FeII/I couples by direct cyclic voltammetry of inducible NOS in organic films on graphite electrodes. These studies allow easy and rapid measurements of electron transfer between the enzyme and the electrode surface and enabled us to detect the interconversion of several coordination states of the enzyme. These studies, coupled with the use of molecular wires as mediators of electron transfer at electrode surfaces, will provide a new way to prove the function of NOS and related enzymes. PUBLICATIONS Hays, A.-M., Dunn, A.R., Chiu, R., Gray, H.B., Stout, C.D. Goodin, D.B. Conformational states of cytochrome P450cam revealed by trapping of synthetic molecular wires. J. Mol. Biol. 344:455, 2004. F i g . 1 . Crystal structure at 1.6 Å of P450cam containing D8A, a synthetic molecular wire. The adamantyl substrate analog is observed at the camphor binding site for wires of different lengths. Changes in the F and G helices in response to wire length illustrate the conformational flexibility in these regions that may be responsible for the diversity of substrate recognition by P450s. Udit, A.K., Belliston-Bittner, W., Glazer, E.C., Nguyen, Y.H.L., Gillon M., Hill, M.G., Marletta, M.A., Goodin, D.B. Gray, H.B. Redox couples of inducible nitric oxide synthase. J. Am. Chem. Soc. 127:11212, 2005. Significant changes in the protein structure near the F and G helices accommodate the changes in linker length. These changes are similar to those that may be responsible for substrate-binding specificity of mammalian P450s and indicate that prokaryotic enzymes have similar conformational flexibility. These changes also suggest the nature of the dynamic intermediates that must exist transiently in solution during substrate S.I. Reed, C. Baskerville, L.-C. Chuang, B. Grünenfelder, M. Henze, J. Keck, V. Liberal, K. Luo, B. Olson, S. Ekholm-Reed, S. Rudyak, O. Sangfelt, A. Smith, C. Spruck, D. Tedesco, F. van Drogen, J. Wohlschlegel, V. Yu Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Control of Cell Division iological processes of great complexity can be approached by beginning with a systematic genetic analysis in which the relevant components are first identified and the consequences of B 224 MOLECULAR BIOLOGY 2005 selectively eliminating the components via mutations are investigated. We use yeast, which is uniquely tractable to this type of analysis, to investigate control of cell division. In recent years, it has become apparent that the most central cellular processes throughout the eukaryotic phylogeny are highly conserved in terms of both the regulatory mechanisms used and the proteins involved. Thus, it has been possible in many instances to generalize from yeast cells to human cells. CONTROL IN YEAST In recent years, we have focused on the role and regulation of the Cdc28 protein kinase (Cdk1). Initially identified by means of a mutational analysis of the yeast cell cycle, this protein kinase and its analogs are ubiquitous in eukaryotic cells and are central to a number of aspects of control of cell-cycle progression. One current area of interest is regulation of cellular morphogenesis by Cdk1. The activity of Cdk1 driven by mitotic cyclins modulates polarized growth in yeast cells. Specifically, these activities depolarize growth by altering the actin cytoskeleton. We found that several proteins that modulate actin structure are targeted by Cdk1, and we are investigating whether these phosphorylation events control actin depolarization. A second major area of interest is the regulation of mitosis. A key aspect of mitotic regulation in yeast is the accumulation of Cdc20, which triggers the transition from metaphase to anaphase. Cdc20 is an essential cofactor of the protein-ubiquitin ligase known as the anaphase-promoting complex or APC/C. It is through the ubiquitin-mediated proteolysis of a specific anaphase inhibitor, securin (Pds1 in yeast), that anaphase is initiated. We found that cells are prevented from entering mitosis when DNA replication is blocked by the drug hydroxyurea, which causes the destabilization of Cdc20 and inhibition of Cdc20 translation. While investigating mitosis, we found that Cks1, a small Cdk1-associated protein, appears to regulate the proteasome. Proteasomes are complex proteases that target ubiquitylated proteins, including important cellcycle regulatory proteins. Surprisingly, we found that Cks1 regulates a nonproteolytic function of proteasomes, the transcriptional activation of Cdc20. Specifically, Cks1 is required to recruit proteasomes to the gene CDC20 for efficient transcriptional elongation. Our investigations of CDC20 led to the conclusion that Cks1 is required for recruitment of proteasomes to and transcriptional elongation of many other genes, as well. Currently, we are elucidating the mechanism whereby Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Cks1 recruits proteasomes and facilitates transcriptional elongation. Our most recent results suggest that Cks1 and proteasomes in conjunction with Cdk1 mediate remodeling of chromatin. CONTROL IN MAMMALIAN CELLS We showed previously that the human homologs of the Cdc28 protein kinase are so highly conserved, structurally and functionally, relative to the yeast protein kinase, that they can function and be regulated properly in a yeast cell. Analyzing control of the cell cycle in mammalian cells, we produced evidence for the existence of regulatory schemes, similar to those elucidated in yeast, that use networks of both positive and negative regulators. A principal research focus is the positive regulator of Cdk2, cyclin E. Cyclin E is often overexpressed and/or deregulated in human cancers. Using a tissue culture model, we showed that deregulation of cyclin E confers genomic instability, probably explaining the link to carcinogenesis. The observation that deregulation of cyclin E confers genomic instability led us to hypothesize a mechanism of cyclin E–mediated carcinogenesis based on accelerated loss of heterozygosity at tumor suppressor loci. We are testing this hypothesis in transgenic mouse models. We showed previously that a cyclin E transgene expressed in mammary epithelium markedly increases loss of heterozygosity at the p53 locus, leading to enhanced mammary carcinogenesis. We are extending these investigations by using mouse prostate, testis, and skin models. In an attempt to understand cyclin E–mediated genomic instability, we are investigating how deregulation of cyclin E affects both S phase and mitosis. Recent data suggest that deregulation of cyclin E impairs DNA replication by interfering with assembly of the prereplication complex. Cyclin E deregulation also impairs the transition from metaphase to anaphase by promoting the accumulation of mitotic checkpoint proteins. Our interest in cyclin E deregulation in cancer led us to examine the pathway for turnover of cyclin E. We showed that phosphorylation-dependent proteolysis of cyclin E depends on a protein-ubiquitin ligase known as SCF hCdc4 . The F-box protein hCdc4 is the specificity factor that targets phosphorylated cyclin E. We are investigating how ubiquitylation of cyclin E is coordinated with other processes required for its degradation. We are also investigating SCFhCdc4 ubiquitylation of other important cellular proteins. Because of the functional relationship between hCdc4 and cyclin E, we are studying the role of muta- MOLECULAR BIOLOGY 2005 tions of hCDC4, the gene that encodes hCdc4, in carcinogenesis. We found that hCDC4 is mutated and most likely is a tumor suppressor in endometrial cancer and breast cancer. In endometrial cancer, tumors with mutations in hCDC4 are more aggressive than tumors without mutations in this gene. Because we showed that loss of hCdc4 leads to deregulation of cyclin E through the cell cycle, these results confirm the observation that in some cancers deregulation of cyclin E is associated with aggressive disease and poor outcome. Another area of interest is the role of Cks proteins in mammals, complementing our research in yeast. Mammals express 2 orthologs of yeast Cks1, known as Cks1 and Cks2. Experiments in mice lacking the gene for Cks1 and Cks2 revealed that each ortholog has a specialized function. Cks1 is required as a cofactor for Skp2-mediated ubiquitylation and turnover of inhibitors p21, p27, and p130. Cks2 is required for the transition from metaphase to anaphase in both male and female meiosis I. Nevertheless, mice nullizygous at the individual loci are viable. However, doubly nullizygous mice have not been observed because embryos die at the morula stage, a finding consistent with an essential redundant function. We found that this function most likely is involved in transcriptional elongation and is linked to chromatin remodeling, as in yeast. PUBLICATIONS Huisman, S.M., Bales, O.A.M., Bertrand, M., Smeets, M.F.M.A., Reed, S.I., Segal, M. Differential contribution of Bud6p and Kar9p in microtubule capture and spindle orientation in S. cerevisiae. J. Cell Biol. 167:231, 2004. Reed, S.I. Cell cycle. In: Cancer: Principles and Practice of Oncology, 7th ed. DeVita V.T., Jr., Hellman, S., Rosenberg, S.A. (Eds.). Lippincott Williams & Wilkins, Philadelphia, 2004, p. 83. Reed, S.I., Rothman, J.H. Cell division, growth and death [editorial]. Curr. Opin. Cell Biol. 16:599, 2004. Spruck C.H., Smith, A.P.L., Ekholm-Reed, S., Sangfelt, O., Keck, J., Strohmaier, H., Méndez, J., Widschwendter, M., Stillman, B., Zetterberg A., Reed, S.I. Deregulation of cyclin E and genomic Instability. In: Hormonal Carcinogenesis IV. Li, J.J., Li, S.A., Llombart-Bosch, A. (Eds.). Springer, New York, 2004, p. 98. Wittenberg, C., Reed, S.I. Cell cycle-dependent transcription in yeast: promoters, transcription factors, and transcriptomes. Oncogene 24:2746, 2005. Wohlschlegel, J.A., Johnson, E.S., Reed, S.I., Yates, J.R. III. Global analysis of protein sumoylation in Saccharomyces cerevisiae. J. Biol. Chem. 279:45662, 2004. Yu, V.P.C.C., Baskerville, C., Grünenfelder, B., Reed, S.I. A kinase-independent function of Cks1 and Cdk1 in regulation of transcription. Mol. Cell 17:145, 2005. Yu, V.P.C.C., Reed, S.I. Cks1 is dispensable for survival in Saccharomyces cerevisiae. Cell Cycle 3:1402, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 225 Regulating Cell Proliferation: Flipping Transcriptional and Proteolytic Switches C. Wittenberg, M. Ashe, R. de Bruin, M. Guaderrama, B.-K. Han, T. Kalashnikova, N. Spielewoy ell proliferation is governed primarily by controlling the activities of positive and negative regulators of cell-cycle transitions. Inhibitors of cyclin-dependent protein kinase (CDK) and the positive regulatory subunits, cyclins, are critical in establishing the proper timing of cell-cycle transitions and in imposing cell-cycle checkpoints. The activities of those proteins are largely regulated via periodic transcriptional activation coupled with regulated proteolysis. We focus primarily on those regulatory mechanisms. As in animal cells, initiation of the cell cycle in the budding yeast Saccharomyces cerevisiae occurs during late G1 phase and is governed by the controlled accumulation of G1 CDK activity. A large family of G1-specific genes, including those for the G1 cyclins Cln1 and Cln2, are coordinately regulated by 2 transcription factors: SBF and MBF. As in metazoans, the transcriptional activation of those genes depends on the activity of a distinct G1 cyclin, Cln3, that acts on promoter-bound transcription factors to promote recruitment of components of the RNA polymerase II complex. By analogy with metazoan Rb, an inhibitor of the E2F transcription factor that is antagonized by cyclin D/CDK, we predicted the existence of a G 1 -specfic transcriptional repressor that is inactivated by Cln3/CDK. Using the combined application of molecular genetics and mass spectrometry–based multidimensional protein identification technology, we identified an SBF-specific transcriptional repressor, Whi5, that is inactivated via phosphorylation by Cln3/CDK. This discovery provides a unifying mechanism for initiation of the cell cycle in yeast and metazoans. We also identified several other transcriptional regulators, including Nrm1, a novel cell cycle–dependent repressor of MBF-dependent transcription. Rather than repressing expression early in the cell cycle as Whi5 does, Nrm1 acts as cells pass into S phase, thereby limiting MBF-dependent gene expression to the G1 phase. Because expression of the gene for NRM1 depends on MBF, the gene cannot act until MBF becomes active. Consequently, the gene confers negative autoregulation C 226 MOLECULAR BIOLOGY 2005 on MBF. Additional factors associated with the 2 transcription factors are under investigation. One of the primary roles of G 1 cyclin-associated CDKs is to promote the ubiquitin-dependent proteolysis of cell-cycle regulators, including the G 1 cyclins themselves. CDK-dependent phosphorylation of a number of proteins targets the proteins for recognition by the Cdc34-SCF ubiquitin ligase complex. Grr1, one of several distinct F box proteins that associate with that complex, confers recognition of specific phosphorylated targets. We are interested in the molecular basis of that recognition. Previously, we showed that the interaction between Grr1 and Cln2 requires basic residues residing in the pocket of the leucine-rich repeat of Grr1 and defined a transferable “degron” in the C terminus of Cln2 that is phosphorylated by the CDK. These findings, combined with our understanding of the mechanisms that govern G1-specific transcription, indicate that an integrated autoregulatory circuit governs the events of G1 phase and ensures the orderly progression of events in the cell cycle. In addition to its role in cell-cycle control, SCFGrr1 plays a central role in regulating the expression of genes induced by glucose and amino acids. We showed that the glucose signal promotes ubiquitin-mediated proteolysis of Mth1, which is required for maintenance of transcriptional repression of glucose-inducible genes. Glucose triggers phosphorylation of Mth1 by casein kinase I, thereby promoting recognition by SCF Grr1 . Surprisingly, recognition of phosphorylated Mth1 requires properties of Grr1 distinct from those required for recognition of phosphorylated G1 cyclins. The same properties are also important for Grr1-dependent recognition of an as yet unknown target required for the activation of amino acid–regulated genes via SPS signaling. Efforts are under way to identify novel targets of Grr1 and to investigate the possibility that Grr1 mediates the coordination of cell-cycle progression with the availability of environmental nutrients. PUBLICATIONS Flick, K., Wittenberg, C. Multiple pathways for suppression of mutants affecting G1-specific transcription in Saccharomyces cerevisiae. Genetics 169:37, 2005. Wittenberg, C. Cell cycle: cyclin guides the way. Nature 434:34, 2005. Wittenberg, C., Reed, S.I. Cell cycle-dependent transcription in yeast: promoters, transcription factors, and transcriptomes. Oncogene 24:2746, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Cell-Cycle Checkpoints, DNA Repair, and Oxidative Stress Response P. Russell, C. Chahwan, S. Coulon, L.-L. Du, P.-H. Gaillard, V. Martin, T. Nakamura, C. Noguchi, E. Noguchi, M. Rodriguez, P. Shanahan, K. Tanaka, H. Zhao he cellular responses to DNA damage and cytotoxic stress are highly conserved through evolution. A fortunate consequence of this conservation is that “simple” eukaryotes such as the fission yeast Schizosaccharomyces pombe can be used as model systems for more complex multicellular organisms. We use S pombe to study cell-cycle checkpoints, DNA repair, and stress response mechanisms. Defects in these mechanisms underlie a number of human diseases, including cancer. T D N A R E P L I C AT I O N C H E C K P O I N T The challenging task of replicating a eukaryotic genome is often made more difficult by conditions that interfere with progression of the replisome, the complex formed by the close association of the key proteins used during DNA replication. Protein complexes bound to DNA, chemical adducts in DNA, and deoxyribonucleotide starvation are among the situations that can impede replisomes. The DNA replication checkpoint senses stalled replication forks and directs cellular responses that help preserve the integrity of the genome. One of these responses is the S-M checkpoint. This checkpoint delays the onset of mitosis (M phase) while DNA synthesis (S phase) is under way, thereby providing time to recover from stalled forks. The same checkpoint also controls how damaged DNA is replicated. DNA-dependent protein kinases, such as ATM and ATR in humans and Rad3 in fission yeast, are central components of the replication checkpoint. Acting in conjunction with regulatory subunits (e.g., Rad26 in fission yeast) and other protein complexes, these kinases activate checkpoint effector kinases. The effector of the replication checkpoint in fission yeast is Cds1 (Chk2). A few years ago, we discovered mediator of replication checkpoint-1 (Mrc1), an adaptor or mediator protein that directs the replication checkpoint signal from Rad3 to Cds1. We recently discovered that the forkhead-associated domain of Cds1 mediates the binding of Cds1 to Mrc1. This interaction allows Rad3 to activate Cds1. MOLECULAR BIOLOGY 2005 Cds1 controls repair systems that are required to tolerate stalled replication forks. We hope to better understand these systems by identifying proteins that associate with the forkhead-associated domain of Cds1. Mus81, a novel protein related to the XPF nucleotide excision repair protein, was identified in a screen for such proteins. We found that Mus81 associates with another protein, Eme1, to form a structure-specific endonuclease that resolves X-shaped Holliday junctions. In recent studies with T. Wang, Stanford University, Stanford, California, and M.N. Boddy, Department of Molecular Biology, we discovered that phosphorylation of Mus81 by Cds1 helps preserve genome integrity when replication forks arrest. We hypothesize that the phosphorylation prevents the Mus81-Eme1 complex from cleaving stalled replication forks. Stalled forks are potentially unstable structures prone to rearrangement and collapse. We previously reported that the protein Swi1 helps preserve stalled forks and is necessary for strong activation of Cds1. Recent studies with J.R. Yates, Department of Cell Biology, indicated that Swi1 associates with Swi3 to form a fork-protection complex. We found that the complex travels with the replisome during DNA replication. It is therefore ideally placed to detect, stabilize, and signal stalled replication forks (Fig. 1). We speculate that Swi1 and Swi3 homologs in humans have equivalent functions. F i g . 1 . Stabilization of stalled replication forks. The fork-protection complex (FPC), which consists of Swi1 and Swi3, travels with the replisome. Mrc1 also appears to travel with the fork. When the replisome stalls at obstructions in the fork or for other reasons, the fork-protection complex and Mrc1 are required for activation of Cds1 by Rad3-Rad26 kinase. The Rad9-Rad1-Hus1 (9-1-1) complex is also required for Cds1 activation. 227 F i g . 2 . The unicellular yeast S pombe divides by medial fission (top left panel). It has 3 chromosomes and approximately 4000 genes. The DNA damage checkpoint arrests division in cells exposed to ionizing radiation (+IR) (top right panel). Pulse-field gel electrophoresis shows that the chromosomes are fragmented by 120 Gy of ionizing radiation (bottom panel). About 3 hours are required to repair the DNA, necessitating a checkpoint that prevents mitosis while DNA repair is under way. is enforced by the protein kinase Chk1, which is activated by Rad3. Activation of Chk1 requires the adaptor protein Crb2. Crb2 is rapidly recruited to double-stranded breaks in DNA. We recently found that Rad3 and Tel1 (the ATM homolog in fission yeast) stimulate Crb2 recruitment by phosphorylating histone H2A at the DNA break site. We also found that the tandem C-terminal BRCT domains in Crb2 are essential for Crb2 homo-oligomerization. Recently, we investigated how Tel1/ATM is recruited for sites of DNA damage and how it is activated. These studies, done in collaboration with T. Hunter, the Salk Institute, La Jolla, California, revealed that Tel1/ATM interacts with the extreme C terminus of Nbs1. Nbs1 is a subunit of the Mre11-Rad50-Nbs1 complex that associates with and processes double-stranded breaks. We found that the interaction with Nbs1 is essential for ATM activation. DNA DAMAGE CHECKPOINT O X I D AT I V E S T R E S S R E S P O N S E The DNA damage checkpoint prevents the onset of mitosis when DNA is damaged (Fig. 2). This checkpoint Oxidative stress caused by reactive oxygen species can be highly toxic, causing damage to proteins, lipids, Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 228 MOLECULAR BIOLOGY 2005 and nucleic acids. Oxidative stress elicits a complex gene expression response that is orchestrated in large part by MAP kinase cascades. The fission yeast Spc1 MAP kinase pathway is homologous to the p38 pathway in humans. We recently discovered Csx1, a protein that collaborates with Spc1 to control gene expression in response to oxidative stress. Csx1 is an RNA-binding protein that mediates global control of gene expression in response to oxidative stress by binding and stabilizing mRNA that encodes Atf1, a transcription factor that is also regulated by Spc1. Most recently, we focused on a newly discovered family of proteins that interact with Csx1. PUBLICATIONS Du, L.L., Moser, B.A., Russell, P. Homo-oligomerization is the essential function of the tandem BRCT domains in the checkpoint protein Crb2. J. Biol. Chem. 279:38409, 2004. Kai, M., Boddy, M.N., Russell, P., Wang, T.S.F. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev. 19:919, 2005. McGowan, C.H., Russell, P. The DNA damage response: sensing and signaling. Curr. Opin. Cell Biol. 16:629, 2004. Nakamura, T.M., Moser, B.A., Du, L.L., Russell, P. Cooperative control of Crb2 by ATM-family and Cdc2 kinases is essential for the DNA damage checkpoint in fission yeast. Mol. Cell. Biol., in press. Noguchi, E., Noguchi, C., McDonald, W.H., Yates, J.R. III, Russell, P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 24:8342, 2004. Tanaka, K., Russell, P. Cds1 phosphorylation by Rad3-Rad26 kinase is mediated by forkhead-associated domain interaction with Mrc1. J. Biol. Chem. 279:32079, 2004. You, Z., Chahwan, C., Bailis, J., Hunter, T. Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25:5363, 2005. Zhao, H., Russell, P. DNA binding domain in the replication checkpoint protein Mrc1 of Schizosaccharomyces pombe. J. Biol. Chem. 279:53023, 2004. DNA Damage Responses in Human Cells C.H. McGowan, V. Blais, H. Gao, E. Langley, A. MacLaren, J. Scorah, E. Taylor omplex multicellular organisms, such as humans, have large numbers of mitotically competent cells that are capable of renewal, repair, and, to some extent, regeneration. The advantages of being able to replace damaged or aged cells are off set by the inherent susceptibility of mitotic cells to acquiring mutations and becoming cancerous. DNA is inherently vulnerable to many sorts of chemical and physical modification; thus, C Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. as they duplicate and divide, cells can acquire mutations. Both spontaneous and induced DNA damage must be repaired with minimal changes if growth, renewal, and repair are to be successful. Our overall objective is to understand how mammalian cells protect themselves from DNA damage and thus from cancer. Eukaryotic cells have evolved with a complex network of DNA repair processes and cell-cycle checkpoint responses that ensure that damaged DNA is repaired before it is replicated and becomes fixed in the genome. These pathways are highly conserved through evolution, and much information about human responses to DNA damage has been gained from studies of simple genetically tractable organisms such as yeast. We use a combination of molecular, cellular, and genetic techniques to determine how these pathways operate in human cells. Checkpoints control the order and timing of events in the cell cycle; they ensure that biochemically independent processes are coupled so that a delay in a critical cell-cycle process will cause a delay in all other aspects of progression of the cycle. In addition, checkpoints coordinate repair with delays in progression of the cell cycle and promote the use of the most appropriate repair pathway. We used genetic models to identify 2 checkpoint kinases in humans that limit progression of the cell cycle when DNA is damaged. One of these kinases, Chk2, is activated in response to DNA damage. Chk2 physically interacts with Mus81-Eme1, a conserved DNA repair protein that has homology to the xeroderma pigmentosum F family of endonucleases. Xeroderma pigmentosum is a cancer-prone disorder that results from a failure to appropriately repair damaged DNA. Biochemical analysis indicates that Mus81-Eme1 has associated endonuclease activity against structurespecific DNA substrates, including Holliday junctions. Enzymatic analysis, immunofluorescence studies, and the use of RNA interference have all contributed to the conclusion that Mus81-Eme1 is required for recombination repair in human cells. We are also using gene targeting to study the function of the Mus81-Eme1 endonuclease in mice. Inactivation of Mus81 in mice increases genomic instability and sensitivity to DNA damage but does not promote tumorogenesis. In addition, we showed that Mus81-Eme1 is specifically required for survival after exposure to cisplatin, mitomycin C, and other commonly used anticancer drugs. As a point of interaction between checkpoint control and DNA repair, the relationship between Mus81-Eme1 and Chk2 most likely will provide information critical to understanding the responses to DNA damage as a whole. MOLECULAR BIOLOGY 2005 229 Anticancer therapy is largely based on the use of genotoxic agents that damage DNA and thus kill dividing cells. Coordination of cell-cycle checkpoints and DNA repair is especially important when unusually high amounts of DNA damage occur after radiation or genotoxic chemotherapy. Hence, a detailed understanding of cellular responses to DNA damage is essential to understanding both the development and the treatment of disease in humans. PUBLICATIONS Dendouga, N., Gao, H., Moechars, D., Janicot, M., Vialard, J., McGowan, C.H. Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol. Cell. Biol. 25:7569, 2005. McGowan, C.H., Russell, P. The DNA damage response: sensing and signaling. Curr Opin. Cell Biol. 16:629, 2004. Zhang, R., Sengupta, S., Yang, Q,, Linke, S.P., Yanaihara, N., Bradsher, J., Blais, V,. McGowan, C.H., Harris, C.C. BLM helicase facilitates Mus81 endonuclease activity in human cells. Cancer Res. 65:2526, 2005. DNA Repair and the Maintenance of Genomic Stability M.N. Boddy, Y. Pavlova, S. Pebenard, G. Raffa NA repair pathways have evolved to protect the genome from ever-present genotoxic agents. Highlighting the importance of the pathways, defects in DNA repair mechanisms strongly predispose the host to cancer and to neurologic and developmental disorders. The DNA repair systems we study in fission yeast are evolutionarily conserved, and therefore our investigations provide a valuable framework for understanding genome maintenance in human cells. Although many DNA repair mechanisms have been described, information on how they are coordinated with necessary changes in chromatin structure is limited. We are studying the essential structural maintenance of chromosomes (SMC) complex Smc5-Smc6. The molecular functions of Smc5-Smc6 are unknown, but the complex is related to the SMC complexes that hold replicated sister chromatids together (cohesin) and condense chromatin before its segregation at mitosis (condensin). In collaboration with J.R. Yates, Department of Cell Biology, we purified the Smc5-Smc6 complex and determined the identity of the core components. The holocomplex consists of the Smc5-Smc6 heterodimer and 6 additional non-SMC elements, Nse1–Nse6 (Fig. 1). We expressed and purified individual components of D Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 1 . Architecture of the Smc5-Smc6 holocomplex. Nse1, Nse3, and Nse4 form a stable heterotrimer that then associates with Smc5. Nse2 interacts directly with Smc5 in the absence of the other Nse proteins. Smc6 interacts directly with Smc5 but none of the other components. Nse5 and Nse6 form a stable heterodimer that also binds directly to Smc5. Double-headed arrows indicate interactions between subcomplexes. Nse5-Nse6 may recruit the holocomplex to stalled replication forks and certain DNA damage sites (black oval on leading-strand template of replication fork). the complex and determined the architecture of the holocomplex. Nse1–Nse4 are essential for growth, and hypomorphic mutants of these proteins cause cellular sensitivity to genotoxic agents such as ultraviolet light and x-rays. Nse5 and Nse6 are nonessential, but cells lacking either protein also are hypersensitive to DNAdamaging agents. Notably, Nse1 and Nse2 contain certain zinc finger domains that implicate these 2 elements in the modification of target proteins with ubiquitin and the small ubiquitin-like protein SUMO. Such protein modifications play roles in DNA repair and chromatin remodeling. Our genetic analyses support a role for the Smc5Smc6 complex in stabilizing replication forks that have stalled at sites of DNA damage. We have also identified a critical role for the Smc5-Smc6 complex in meiosis, the process that generates gametes for reproduction and genetic diversity. A critical feature of meiosis is the programmed formation of DNA double-strand breaks followed by repair of the breaks via homologous recombination. We found that the Smc5-Smc6 complex functions in the correct repair of the breaks and that mutants 230 MOLECULAR BIOLOGY 2005 of Smc5-Smc6 do not segregate homologous chromosomes at the first meiotic division. Finally, we identified a physical interaction between the Smc5-Smc6 complex and Rad60, an essential DNA repair factor required for the homologous recombination repair of DNA. Rad60 is regulated by the replication checkpoint, and thus we can study the important but poorly defined interface between DNA repair and cell-cycle checkpoints. PUBLICATIONS Kai, M., Boddy, M.N., Russell, P., Wang, T.S. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev. 19:919, 2005. Pebernard, S., McDonald, W.H., Pavlova, Y., Yates, J.R., III, Boddy, M.N. Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol. Biol. Cell 15:4866, 2004. Delineation of Oncogenic and Tumor-Suppressing Pathways via Genetic Approaches P. Sun, Q. Deng, C. Kannemeier, R. Liao, B. Moser ur major interests are the genetic alterations involved in tumorigenesis and the cellular pathways that must be altered during oncogenic transformation. To this end, we analyzed the behaviors of primary, normal human cells after stable transduction of oncogenes, such as ras and MPM2. Members of the ras family of oncogenes encode small GTP-binding proteins that transduce growth signals. Constitutive activation of ras often occurs in tumors and contributes to tumor development. In normal cells, activation of ras triggers an antioncogenic response called premature senescence, a stable growth arrest that must be overcome before transformation occurs. We showed that ras induces senescence through sequential activation of 2 MAP kinase pathways. Initially, ras activates the MAP kinase kinase (MEK)–extracellular signal–regulated kinase (ERK) pathway. Sustained activation of MEK-ERK turns on the stress-induced p38 pathway, which subsequently causes senescence. These results revealed a novel, tumor-suppressing function of p38, in addition to its known roles in inflammation and stress responses. In other studies, we identified additional signaling components, either upstream or downstream of p38, that mediate premature senescence. To determine how premature senescence is bypassed in tumors, we dissected the functions of an adenovi- O Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. rus-encoded oncoprotein, E1A, that can rescue cells from ras-induced senescence. E1A directly binds to and inhibits the functions of several cellular proteins, such as members of the Rb family, p300/CBP, and p400, that have been implicated in tumor-suppressing pathways. Our results indicated that senescence-bypassing activity resides in the N terminus of E1A and requires binding of both Rb and p300/CBP, but not binding of p400. Although interference with the p16 INK4A /Rb pathway or with p300/CBP functions alone did not result in bypassing of senescence, these 2 types of genetic alterations complemented mutants of E1A with defects in Rb binding and p300/CBP binding, respectively, to rescue cells from ras-induced senescence and lead to cellular transformation. Therefore, genetic alterations that disrupt the p16INK4A/Rb pathway and those that perturb the p300/CBP functions cooperate to bypass ras-induced senescence. These results indicate that p300 and CBP are integral components of the senescence pathway. Both p300 and CBP have tumorsuppressing functions. The critical role of p300 and CBP in the senescence response has provided a mechanistic basis for the tumor-suppressing function of these proteins. Another focus of our research is MDM2, an oncogene that can mediate transformation primarily through inactivation of the tumor suppressor protein p53. However, we found that MDM2 confers resistance to a growth-inhibitory cytokine, transforming growth factor β, through a p53-independent mechanism. Currently, we are delineating this p53-independent activity of MDM2, which may play an important role in tumorigenesis. In other studies, we are systematically searching for genetic alterations that contribute to specific tumorassociated phenotypes, such as drug resistance, cellular immortalization, and metastasis. For these investigations, we are using cDNA expression libraries or libraries of short interfering RNAs. PUBLICATIONS de Parseval, A., Chatterji, U., Morris, G., Sun, P., Elder, J.H. Fine mapping of CD134 residues critical for interaction with feline immunodeficiency virus. Nat. Struct. Mol. Biol. 12:60, 2005. de Parseval, A., Chatterji, U., Sun, P., Elder, J.H. Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc. Natl. Acad. Sci. U. S. A. 101:13044, 2004. de Parseval, A., Ngo, S., Sun, P., Elder, J.H. Factors that increase the effective concentration of CXCR4 dictate feline immunodeficiency virus tropism and kinetics of replication. J. Virol. 78:9132, 2004. Deng, Q., Li, Y., Tedesco, D., Liao, R., Fuhrmann, G., Sun, P. The ability of E1A to rescue ras-induced premature senescence and confer transformation relies on inactivation of both p300/CBP and Rb family proteins. Cancer Res. 65:8298, 2005. MOLECULAR BIOLOGY 2005 The 5-HT7 Receptor as a Target in Depression and Schizophrenia P.B. Hedlund, P.E. Danielson, S. Huitrón-Reséndiz, S.J. Hendriksen, S. Semenova, M.A. Geyer, A. Markou, J.G. Sutcliffe erotonin (5-HT) is produced by a small group of nuclei in the brain stem that send their projections to a vast number of receptive fields. The family of receptors for 5-HT is the most diverse family that binds a single ligand; it has at least 14 members. One of these is the 5-HT7 receptor, which we previously discovered. In earlier studies, we showed that this receptor mediates resetting of circadian rhythms by the hypothalamus. Despite vast differences in amino acid sequence between the 5-HT 7 receptor and the 5-HT1A receptor, the 2 share considerable pharmacology and have been implicated in some of the same functions. 5-HT1A is more abundant than 5-HT7, but the areas of the brain that express the 2 receptors overlap considerably. We produced mutant mice in which the gene for the 5-HT7 receptor was inactivated. Studies with these mice and SB-266970, a 5-HT7-selective antagonist, indicated that this receptor mediates serotonin-induced hypothermia and is important for fine tuning of temperature homeostasis. Sleep, circadian rhythm, and mood are related phenomena. 5-HT7-selective antagonists increase REM sleep latency and decrease the cumulative duration of REM sleep, patterns the opposite of those found in patients with clinical depression. Several antidepressants activate 5-HT7 neurons in the circadian control area of the hypothalamus, and chronic treatment with antidepressants diminishes both activation and 5-HT 7 binding there. We examined sleep parameters in the mutant mice in which the gene for the 5-HT 7 receptor was inactivated. We found that they spent less time than normal mice in REM sleep. This pattern is the opposite of that found in humans with depression. Two models of behavioral despair, the forced swim test and the tail suspension test, make rats and mice immobile. This immobility, or helplessness, is likened to depression in humans because a high correlation exists between the ability of antidepressant drugs to reverse immobility in rodents and to be effective clinically in humans. Furthermore, mice selectively bred to have increased helplessness in these behavioral despair S Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 231 tests resemble patients with clinical depression. The mice have decreased REM latency and more cumulative REM sleep, elevated levels of corticosterone, a decreased 5-HT metabolism index, and altered serotonin-induced hypothermia. We examined unmedicated 5-HT7 mutant mice in these tests and found that the mice remained significantly more mobile than unmedicated normal mice during both the forced swim and the tail suspension tests. Normal mice medicated with the 5-HT7-selective antagonist SB-266970 mimicked the mobility of unmedicated mutant mice, whereas the selective antagonist had no effect on the mobility of mutant mice. A selective serotonin reuptake inhibitor increased mobility in both types of mice (albeit at a lower concentration in the mutant mice), suggesting that the inhibitor worked through an independent mechanism. These results are consistent with the notion that the 5-HT7 mutant mice have characteristics of a partially “antidepressed” state: they spend less time in REM sleep, have reduced immobility in the forced swim and tail suspension tests, and have decreases in serotonin-induced hypothermia. Normal mice medicated with 5-HT7-selective antagonists resemble unmedicated 5-HT7 mutant mice in these measures. These findings suggest that 5-HT7-selective antagonists might be sufficient treatment for some aspects of clinical depression. Several antipsychotic drugs have high affinity for the 5-HT7 receptor. We examined the role of 5-HT7 receptors in an animal model of schizophrenia: phencyclidine-induced disruption of prepulse inhibition of the acoustic startle reflex. In untreated mice, we found no difference between mice in which the gene for the 5-HT7 receptor was inactivated and wild-type mice in startle response or in prepulse inhibition regardless of prepulse intensity, interstimulus interval, or pulse intensity. SB-269970 had no effect on prepulse inhibition. The disruption of prepulse inhibition produced by phencyclidine in wild-type mice did not occur in the mutant mice. Similarly, the effect of phencyclidine on prepulse inhibition was reduced by SB-269970 in wild-type mice. The results indicate a specific role for the 5-HT7 receptor in the glutamatergic prepulse inhibition model of schizophrenia. PUBLICATIONS de Lecea, L., Sutcliffe, J.G. Hypocretin as a wakefulness regulatory peptide. In: The Orexin/Hypocretin System: Physiology and Pathophysiology. Nishino, S., Sakurai, T. (Eds.). Humana Press, Totowa, NJ, 2005, p. 143. A volume in the series Contemporary Clinical Neuroscience. de Lecea, L., Sutcliffe, J.G. (Eds.). The Hypocretins: Integrators of Physiological Functions. Plenum Press, New York, 2005. 232 MOLECULAR BIOLOGY 2005 Hedlund, P.B., Huitrón-Reséndiz, S., Henriksen, S.J., Sutcliffe, J.G. 5-HT7 receptor inhibition and inactivation induce antidepressantlike behavior and sleep pattern. Biol. Psychiatry, in press. Sutcliffe, J.G., de Lecea, L. Hypocretins/orexins in brain function. In: Handbook of Neurochemistry and Molecular Neurobiology. Lim, R. (Ed.). Springer, New York, in press. changes in expression were detected in any of the genes tested. These findings indicate that mutant huntingtin protein causes selective deficits in the expression of mRNAs responsible for striatum-specific physiologic changes. Furthermore, the results suggest that although both Huntington’s disease and Parkinson’s disease involve striatal dysfunction, the differences in the molecular pathologic changes associated with the 2 diseases are distinct. Sutcliffe J.G., de Lecea, L. Not asleep, not quite awake. Nat. Med. 10:673, 2004. MOLECULAR MARKERS OF SCHIZOPHRENIA Hedlund, P.B., Sutcliffe, J.G. Functional, molecular and pharmacological advances in 5-HT7 receptor research. Trends Pharmacol. Sci. 25:481, 2004. Hedlund, P.B., Sutcliffe, J.G. 5-HT7 receptors as favorable pharmacological targets for drug discovery. In: The Serotonin Receptors: From Molecular Pharmacology to Human Therapeutics. Roth, B.L. (Ed.). Humana Press, Totowa, NJ, in press. Sutcliffe, J.G., de Lecea, L. The hypocretin/orexin system. In: Handbook of Contemporary Neuropharmacology. Sibley, D. (Ed.). Wiley & Sons, Hoboken, NJ, in press. Ziolkowska, B., Gieryk, A., Bilecki, W., Wawrzczak-Bargiela, A., Wedzony, K., Chocyk, A., Danielson, P.E., Thomas, E.A., Hilbush, B.S., Sutcliffe, J.G., Przewlocki, R. Regulation of α-synuclein expression in limbic and motor brain regions of morphine-treated mice. J. Neurosci. 25:4996, 2005. Molecular Neurobiology of CNS Disorders E.A. Thomas, J.G. Sutcliffe, P.A. Desplats, S. Narayan, K.E. Kass, W. Huang G E N E E X P R E S S I O N I N S T R I ATA L D I S O R D E R S e have identified and cataloged approximately 50 genes that are predominantly expressed in the striatum in the brain. Our long-standing hypothesis is that such genes most likely encode proteins that are preferentially associated with particular physiologic processes in the striatum and therefore may be relevant to striatal disorders. Using oligonucleotide microarrays, we measured expression of these genes simultaneously in the striatum of R6/1 mice, a transgenic model of Huntington’s disease. A total of 81% of striatal genes had increased expression in mice in presymptomatic and/or symptomatic stages of illness. Changes in expression of genes associated with G protein signaling and calcium homeostasis are of particular interest for future studies. The most striking decrease occurred in β4, a newly identified subunit of the sodium channel. Changes in expression began when the mice were 8 weeks old, and expression had progressively decreased almost 10-fold by the time the mice were 8 months old. Two novel sequences with highly specific striatal expression also had differences in expression throughout the life span of the mutant mice, as determined by in situ hybridization analysis. Expression differences of 15 of the striatum-enriched genes were tested in rats treated with 6-hydroxydopamine, a rodent model of Parkinson’s disease. No W Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Schizophrenia is a life-long mental illness with variable expression and unknown etiology. The major clinical manifestations of schizophrenia at the time of onset of the illness are psychotic symptoms; however, as the illness progresses, the negative symptoms become more predominant. In addition, many other neurologic aspects change during the course of the illness. We are interested in the molecular factors that influence manifestation of the symptoms and the course of schizophrenia after its onset and how treatment modifies the effects of illness. Using oligonucleotide microarrays, we generated gene expression profiles from tissue samples obtained at autopsy from the prefrontal cortex of patients with schizophrenia of short and long duration. Because correct treatment early in the illness is thought to have a beneficial effect on the outcome of schizophrenia, the identification of genes involved in the early and late stages of disease will be important for understanding the progression of the illness. PUBLICATIONS Dean, B., Keriakous, D., Thomas, E.A., Scarr, E. Understanding the pathology of schizophrenia: the impact of high-throughput screening of the genome and proteome in postmortem CNS. Curr. Psychiatry Rev. 1:1, 2005. Digney, A., Keriakous, D., Scarr, E., Thomas, E.A., Dean, B. Differential changes in apolipoprotein E in schizophrenia and bipolar I disorder. Biol. Psychiatry 57:711, 2005. Yao, J.K., Thomas, E.A., Reddy, R.D., Keshavan, M.S. Association of plasma apolipoproteins D with RBC membrane arachidonic acid levels in schizophrenia. Schizophr. Res. 72:259, 2005. Ziolkowska, B., Gieryk, A., Bilecki, W., Wawrzczak-Bargiela, A., Wedzony, K., Chocyk, A., Danielson, P.E., Thomas, E.A., Hilbush, B.S., Sutcliffe, J.G., Przewlocki, R. Regulation of α-synuclein expression in limbic and motor brain regions of morphine-treated mice. J. Neurosci. 25:4996, 2005. MOLECULAR BIOLOGY 2005 Molecular Biology of Sleep L. de Lecea, C. Suzuki, C. Pañeda, B. Boutrel,* R. WinskySommerer, A. Coda, S. Huitrón-Reséndiz,* A.J. Roberts,* J.G. Sutcliffe, G.F. Koob,* S.J. Henriksen* * Molecular and Integrative Neurosciences Department, Scripps Research ur goal is to understand the cellular and molecular components that modulate cortical activity and sleep. In particular, we focus on the characterization of neuropeptides first described by our group: cortistatin and the hypocretins. O C O R T I S TAT I N Cortistatin is a neuropeptide expressed in the cerebral cortex. Of its 14 residues, 11 also occur in the neuropeptide somatostatin. However, cortistatin and somatostatin have different physiologic functions. Cortistatin is neuroinhibitory and promotes sleep. We generated mice deficient in cortistatin and determined their behavioral profile in collaboration with A.J. Roberts, Molecular and Integrative Neurosciences Department. Because cortistatin has anticonvulsant activity, we tested seizure susceptibility in cortistatindeficient mice. We also did gene array studies to determine the consequences of cortistatin deficiency in mice lacking the gene for this neuropeptide. Our results suggest that cortistatin has multiple functions in the maintenance of cortical excitability. THE HYPOCRETINS The hypocretins, 2 neuropeptides derived from the same precursor, are produced in a few thousand cells in the lateral part of the hypothalamus. The hypocretins are key molecules for the stability of the states of vigilance. Lack of hypocretin peptides or hypocretin-producing neurons produces narcolepsy, a sleep disorder characterized by uninvited intrusions of sleep into wakefulness. Patients with narcolepsy experience excessive daytime sleepiness and cataplexy, a sudden loss of muscle tone upon certain stimuli. Recent studies indicated that patients with narcolepsy lack hypocretin-expressing cells, suggesting that narcolepsy is a neurodegenerative disease of the hypocretinergic system. In anatomic and electrophysiologic experiments, we found that neurons expressing hypocretin are contacted by neurons expressing corticotropin-releasing factor (CRF), a major component of the stress response. Hypocretin neurons contain CRF receptors. Intracellular recordings in hypothalamic slices from transgenic mice that express green fluorescent protein under the Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 233 control of the hypocretin promoter indicated that CRF depolarizes hypocretin neurons through the CRF 1 receptor. Further, hypocretin neurons are not activated upon stress in mice that lack the gene for this receptor. These data suggest a close association between the CRF and hypocretin systems in the acute stress response. Because CRF is involved in addiction and because hypocretin neurons project to key areas involved in brain reward, we hypothesized that hypocretin neurons might be involved in addiction-related behaviors. We found that hypocretin-1 leads to the reinstatement of previously extinguished cocaine-seeking behavior but does not alter cocaine intake in rats. In collaboration with P.J. Kenny and A. Markou, Molecular and Integrative Neurosciences Department, we discovered that hypocretin-1 negatively regulates the activity of brain reward circuitries. Hypocretin-induced reinstatement of cocaine seeking can be prevented by simultaneous blockade of noradrenergic and CRF systems but not by blockade of either system alone. These findings reveal a previously unidentified role for hypocretins in drug craving and relapse behavior. Moreover, hypocretins may drive drug seeking through induction of a negative affective state by activation of stress pathways in the brain. NEUROPEPTIDE S Neuropeptide S is a newly discovered neuropeptide expressed prominently in a few hundred neurons in the area near the locus coeruleus. We found that infusion of neuropeptide S into the brain ventricles in mice dramatically enhanced wakefulness and suppressed anxiety. The neuropeptide activated several brain nuclei related to arousal. We showed that neurons expressing neuropeptide S project to and depolarize neurons expressing hypocretin. Our data strongly suggest that neuropeptide S is an important modulator of sleep and waking. PUBLICATIONS de Lecea, L. Reverse genetics and the study of sleep. In: Sleep: Circuits and Functions. Luppi, P.-H. (Ed.). CRC Press, Boca Raton, FL, 2004, p. 109. de Lecea, L., Sutcliffe, J.G. The hypocretins and sleep. FEBS J., in press. de Lecea, L., Sutcliffe, J.G. (Eds.) Hypocretins: Integrators of Physiological Functions. Springer, New York, 2005. Huitrón-Reséndiz, S., Kristensen, M.P., Sánchez-Alavez, M., Clark, S.D., Grupke, S.L., Tyler, C., Suzuki, C., Nothacker, H.P., Civelli, O., Criado, J.R., Henriksen, S.J., Leonard, C.S., de Lecea, L. Urotensin II modulates rapid eye movement sleep through activation of brainstem cholinergic neurons. J. Neurosci. 25:5465, 2005. Levine, A.S., Winsky-Sommerer, R., Huitrón-Reséndiz, S., Grace, M.K., de Lecea, L. Injection of neuropeptide W into paraventricular nucleus of hypothalamus increases food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288:R1727, 2005. Martin, G., Guadaño-Ferraz, A., Morte, B., Ahmed, S., Koob, G.F., de Lecea, L. Siggins, G.R. Chronic morphine treatment alters N-methyl-D-aspartate receptors in freshly isolated neurons from nucleus accumbens. J. Pharmacol. Exp. Ther. 311:265-73, 2004. 234 MOLECULAR BIOLOGY 2005 Pañeda, C., Winsky-Sommerer, R., Boutrel, B., de Lecea, L. The corticotropinreleasing factor-hypocretin connection: implications in stress response and addiction. Drug News Perspect. 18:250, 2005. Spier, A.D., Fabre, V., de Lecea, L. Cortistatin radioligand binding in wild-type and somatostatin receptor-deficient mouse brain. Regul. Pept. 124:179, 2005. Sutcliffe, J.G., de Lecea L. Not asleep, not quite awake. Nat. Med. 10:673, 2004. Tallent, M.K., Fabre, V., Qiu, C., Calbet, M., Lamp, T., Baratta, M.V., Suzuki, C., Siggins, G.R., Henriksen, S.J., Criado, J.R., Roberts, A., de Lecea, L., Cortistatin overexpression in transgenic mice produces deficits in synaptic plasticity and learning. Mol. Cell. Neurosci., in press. Ureña, J.M., La Torre, A., Martínez, A., Lowenstein, E., Franco, N., Winsky-Sommerer, R., Fontana, X., Casaroli-Marano, R., Ibáñez-Sabio, M.A., Pascual, M., del Rio, J.A., de Lecea, L., Soriano, E. Expression, synaptic localization, and developmental regulation of Ack1/Pyk1, a cytoplasmic tyrosine kinase highly expressed in the developing and adult brain. J. Comp. Neurol. 490:119, 2005. Winsky-Sommerer, R., Boutrel, B., de Lecea , L. Stress and arousal: the corticotropin-releasing factor/hypocretin circuitry. J. Mol. Neurobiol., in press. Winsky-Sommerer, R., Yamanaka, A., Diano, S., Borok, E., Roberts, A., Sakurai, T., Kilduff, T.S., Horvath, T.L., de Lecea, L. Interaction between the corticotropinreleasing factor system and hypocretins (orexins): a novel circuit mediating stress response. J. Neurosci. 24:11439, 2004. Xu, Y., Reinscheid, R.R., Huitrón-Reséndiz, S., Clark, S.D., Wang, Z., Lin, S.H., Brucher, F.A., Zeng, J., Ly, H.K., Henriksen, S.J., de Lecea, L., Civelli, O. Neuropeptide S: a novel neuropeptide promoting arousal and anxiolytic-like effects. Neuron 43:487, 2004. Molecular Neuroscience: Lysophospholipid Signaling, Neural Aneuploidy J. Chun, B. Almeida, B. Anliker, E. Birgbauer, M. Fontanoz, S. Gardell, C. Paczkowski, D. Herr, D. Kaushal, G. Kennedy, M. Kingsbury, C.W. Lee, M. McConnell, M. McCreight, S. Peterson, S. Rehen, R. Rivera, M. Lu, W. Westra, A.H. Yang, X.Q. Ye, Y. Yung, L. Zhu nderstanding the nervous system—how it arises developmentally and how it carries out its myriad complex tasks in normal and diseased states— is a major challenge. We are studying 2 topics with both basic and potentially therapeutic relevance: the role of lysophospholipid signaling and the role of genomic alterations within individual neurons as manifested by aneuploidy. U LY S O P H O S P H O L I P I D S I G N A L I N G Lysophospholipids are simple phospholipids containing a glycerophosphate or glycerosphingoid backbone and single acyl chain of varied length and saturation. Two major forms of lysophospholipids are lysophosphatidic acid and sphingosine 1-phosphate (Fig. 1). It is now clear from our research and that of many othPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 1 . Chemical structures of lysophosphatidic acid and sphin- gosine 1-phosphate. ers that most important actions of lysophospholipids are mediated by cognate G protein–coupled receptors. A growing range of neurobiological functions is being identified, particularly effects on Schwann cells and oligodendrocytes, which are involved in myelination, and on neuroprogenitor cells of the cerebral cortex. To determine receptor selectivity and actual neurobiological function, we are producing mice that lack the genes for single and multiple receptors. In collaboration with other scientists at Scripps Research, we are developing chemical tools to dissect the in vivo function of lysophosphatidic acid and sphingosine 1-phosphate. During 2004, the range of new biological functions for receptor-mediated lysophospholipid signaling continued to grow. With collaborators from around the world, we showed that lysophospholipid signaling influences the cardiovascular system, the immune system, cancer cell motility, and, especially, neuropathic pain and multiple sclerosis. Neuropathic pain is pain due to nerve damage or dysfunction. Mechanisms for the initiation of this type of pain are poorly understood. In a murine model of neuropathic pain, activation of a single lysophospholipid receptor was necessary for the initiation of pain; such pain did not develop in mice that lacked the gene for the receptor. Another medically important disease, multiple sclerosis, can be approximated in animals by immunization with myelin antigens to produce experimental autoimmune encephalomyelitis. Agonists for lysophospholipid receptors (specifically, sphingosine 1-phosphate receptor agonists) abrogated the disability normally produced by experimental autoimmune encephalomyelitis, suggesting a role for this signaling pathway in the medical biology and a possible therapy MOLECULAR BIOLOGY 2005 for multiple sclerosis. We are expanding these themes in previously identified and new biological systems. 235 Baudhuin, L.M., Jiang, Y., Zaslavsky, A., Ishii, I., Chun, J., Xu, Y. S1P3-mediated Akt activation and cross-talk with platelet-derived growth factor receptor (PDGFR). FASEB J. 18:341, 2004. NORMAL NEURAL ANEUPLOIDY Are all neurons of the brain genetically identical, as is widely assumed, or are differences encoded within individual genomes? Using a combination of spectral karyotyping, which “paints” chromosomes to allow their unambiguous detection, and fluorescence in situ hybridization, which uses labeled point-probes to identify discrete genetic loci in interphase cells, we detected a substantial degree of genomic variation in the normal brain. During neurogenesis, approximately one third of all cells are aneuploid, produced, at least in part, by chromosome missegregation mechanisms. In postmitotic neurons, in which spectral karyotyping cannot be used because neurons are in interphase, fluorescence in situ hybridization of sex chromosomes revealed a high percentage of aneuploidy, and the total number of aneuploid cells is certainly higher if the remaining autosomes are considered (Fig. 2). Chun, J. Choices, choices, choices. Nat. Neurosci. 7:323, 2004. Girkontaite, I., Sakk, V., Wagner, M., Borggrefe, T., Tedford, K., Chun, J., Fischer, K.-D. The sphingosine-1-phosphate (S1P) lysophospholipid receptor S1P3 regulates MAdCAM-1+ endothelial cells in splenic marginal sinus organization. J. Exp. Med. 200:1491, 2004. Hama, K., Aoki, J., Fukaya, M., Kishi, Y., Sakai, T., Suzuki, R., Ohta, H., Yamori, T., Watanabe, M., Chun, J., Arai, H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J. Biol. Chem. 279:17634, 2004. Inoue, M., Rashid, M.H., Fujita, R., Contos, J.J., Chun, J., Ueda, H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling [published correction appears in Nat. Med. 10:755, 2004]. Nat. Med. 10:712, 2004. Ishii, I., Fukushima, N., Ye, X., Chun, J. Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 73:321, 2004. Kingsbury, M.A., Rehen, S.K., Ye, X., Chun, J. Genetics and cell biology of lysophosphatidic acid receptor-mediated signaling during cortical neurogenesis. J. Cell. Biochem. 92:1004, 2004. Levkau, B., Hermann, S., Theilmeier, G., van der Giet, M., Chun, J., Schober, O., Schäfers, M. High-density lipoprotein stimulates myocardial perfusion in vivo. Circulation 110:3355, 2004. McConnell, M.J., Kaushal, D., Yang, A.H., Kingsbury, M.A., Rehen, S.K., Treuner, K., Helton, R., Annas, E.G., Chun, J., Barlow, C. Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in ATM-deficient but not the Trp53-deficient adult cerebral cortex. J. Neurosci. 24:8090, 2004. Nofer, J.-R., van der Giet, M., Tölle, M., Wolinska, I., von Wnuck-Lipinski, K., Baba, H.A., Gödecke, A., Tietge, U.J., Ishii, I., Kleuser, B., Schäfers, M., Fobker, M., Zidek, W., Assmann, G., Chun, J., Levkau, B. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Invest. 113:569, 2004. Rao, T.S., Lariosa-Willingham, K.D., Lin, F.-F. Yu, N., Tham, C.-S., Chun, J., Webb, M. Growth factor pre-treatment differentially regulates phosphoinositide turnover downstream of lysophospholipid receptor and metabotropic glutamate receptors in cultured rat cerebrocortical astrocytes. Int. J. Dev. Neurosci. 22:131, 2004. F i g . 2 . Examples of neural aneuploidy in different regions of the brain in adult mice as revealed by fluorescence in situ hybridization. During 2004, by analyzing mice deficient in DNA surveillance or repair molecules, we detected a new influence on the generation of aneuploidy. One of these molecules, the mutated protein ATM, is the cause of the rare genetic disease ataxia-telangiectasia. Elimination of the gene for ATM or the gene for XRCC5, another molecule involved in DNA surveillance and repair, resulted in major increases in the number and severity of aneuploid neural progenitor/stem cells, indicating a positive biological link between aneuploidy and molecules involved with genome integrity. Currently, we are exploring the basic phenomenologic aspects and functional importance of neural aneuploidy during development and in disease processes. PUBLICATIONS Anliker, B., Chun, J. Cell surface receptors in lysophospholipid signaling. Semin. Cell Dev. Biol. 15:457, 2004. Anliker, B., Chun, J. Lysophospholipid G protein-coupled receptors. J. Biol. Chem. 279:20555, 2004. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. Sanna, M.G., Liao, J., Jo, E., Alfonso, C., Ahn, M.Y., Peterson, M.S., Webb, B., Lefebvre, S., Chun, J., Gray, N., Rosen, H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 279:13839, 2004. Webb, M., Tham, C.-S., Lin, F.-F., Lariosa-Willingham, K., Yu, N., Hale, J., Mandala, S., Chun, J., Rao, T.S. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J. Neuroimmunol. 153:108, 2004. Chemical Glycobiology in the Immune System J.C. Paulson, P. Bengtson, O. Blixt, B.E. Collins, S. Han, T. Islam, H. Tateno, Q. Yan e investigate the roles of glycan-binding proteins that mediate cellular processes central to immunoregulation and human disease. We work at the interface of biology and chemistry to understand how the interaction of glycan-binding proteins with their ligands modulates the functions of the pro- W 236 MOLECULAR BIOLOGY 2005 teins in cell-cell adhesion and cell signaling. Projects fall into 2 main areas: (1) functions of glycan-binding proteins expressed on leukocytes and (2) regulation of the synthesis of the carbohydrate ligands of the proteins during leukocyte activation and differentiation. Our multidisciplinary approach is complemented by a diverse group of chemists, biochemists, cell biologists, and molecular biologists. sure to ultraviolet light (Fig. 1). The striking finding is that microdomain localization of CD22, not glycan structure alone, strongly influences the glycoprotein ligands CD22 interacts with, providing insights into how glycan ligands influence the function of this molecule. This basic observation on siglec-ligand interactions most likely is recapitulated by other members of the siglec family. S I G L E C FA M I LY O F C E L L A D H E S I O N P R O T E I N S A total of 11 human and 8 mouse siglecs have been identified so far, and most siglecs are expressed on leukocytes. The siglecs are a subfamily of the immunoglobulin superfamily. They have variable numbers of extracellular Ig domains, including a unique, homologous N-terminal Ig domain that confers the ability to bind to sialic acid–containing carbohydrate groups (sialosides) of glycoproteins and glycolipids. The cytoplasmic domains of the siglecs typically contain one or more immunoreceptor tyrosine-based inhibitory motifs characteristic of accessory proteins that regulate transmembrane signaling of cell-surface receptor proteins. To dissect the biology of the siglecs, we use novel carbohydrate probes that modulate the function of the proteins. We use chemoenzymatic approaches to synthesize sialoside analogs recognized by siglecs. The analogs range from potent inhibitors to multivalent probes of siglec binding to monovalent sialic acid analogs that can be fed to cells and incorporated into cell-surface glycoproteins to add chemical functionality or alter the affinity of sialoside ligands for cell-surface siglecs. Projects on several members of the siglec family are ongoing. CD22 (siglec-2) is an accessory molecule of the B-cell receptor complex; it has both positive and negative effects on receptor signaling. The carbohydrate ligand recognized by CD22 is the sequence sialic acid α-2-6-galactose, which commonly terminates N-linked carbohydrate groups of glycoproteins. Significantly, ablation of the gene that encodes β-galactoside α-2,6-sialyltransferase I, the enzyme responsible for synthesis of this carbohydrate in mice, causes a marked deficiency in antibody production in response to vaccination with T cell–dependent or T cell–independent antigens, establishing the importance of the ligand in CD22 function. We developed a novel method for in situ photoaffinity cross-linking of CD22 to its ligands on the same cell (cis) or on an adjacent cell (trans); we use a 9-arylazide-sialic acid that is taken up by cells and incorporated into cell-surface glycoproteins, allowing the glycoproteins to be cross-linked to CD22 upon expoPublished by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. F i g . 1 . Bioengineering of cell-surface glycoproteins to carry 9-aryl- azide-sialic acids for in situ photoaffinity cross-linking of CD22 to its ligands. Other members of the siglec family differ from CD22 both in cellular distribution and in specificity for recognition of sialic acid–containing oligosaccharides. We are evaluating the roles of siglec-7 and siglec-9 in regulation of human T-cell receptor signaling, the role of siglec-F in the biology of eosinophils in mice, and the role of myelin-associated glycoprotein (siglec-4) in regulation of neurite formation. We recently developed a novel approach in which a robotically printed glycan array is used for combinatorial assessment of the effects of sialoside analogs on the affinity of siglecs. This method should be a rapid one for developing high-affinity sialoside probes for each of the siglecs to facilitate investigations into siglec biology. R E G U L AT I O N O F L E U K O C Y T E G LY C O S Y L AT I O N Activation of lymphocytes and other leukocytes induces programmed changes in glycosylation. Such changes regulate leukocyte trafficking and can modulate the functions of carbohydrate-binding proteins. We are systematically investigating the changes in glycosylation that occur in B and T lymphocytes after activation in order to elucidate the underlying molecular mechanisms for these changes and their biological relevance. To this end, in collaboration with S. Head and the Consortium for Functional Glycomics (http://www .functionalglycomics.org), we participated in the development and use of a custom microarray of glycosyl- MOLECULAR BIOLOGY 2005 transferase genes, and in collaboration with A. Dell, Imperial College London, London, England, we correlated dramatic changes in gene expression with changes in the glycan profiles of the resting and activated B and T cells. PUBLICATIONS Amado, M., Yan, Q., Comelli, E.M., Collins, B.D. Paulson, J.C. Peanut agglutinin high phenotype of activated CD8+ T cells results from de novo synthesis of CD45 glycans. J. Biol. Chem. 279:36689, 2004. Blixt, O., Head, S., Mondala, T., Scanlan, C., Huflejt, M.E., Alvarez, R., Bryan, M.C., Fazio, F., Calarese, D., Stevens, J., Razi, N., Stevens, D.J., Skehel, J.J., van Die, I., Burton, D.R., Wilson, I.A., Cummings, R., Bovin, N., Wong, C-H., Paulson, J.C. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. U. S. A. 101:17033, 2004. Blixt, O., Vasiliu, D., Allin, K., Jacobsen, N., Warnock, D., Razi, N., Paulson, J.C., Bernatchez, S., Gilbert, M., Wakarchuk, W. Chemoenzymatic synthesis of 2-azidoethyl-ganglio-oligosaccharides GD3, GT3, GM2, GD2, GT2, GM1, and GD1a. Carbohydr. Res. 340:1963, 2005. Bryan, M.C., Fazio, F., Lee, H.-K., Huang, C.-Y., Chang, A., Best, M.D., Calarese, D.A., Blixt, O., Paulson, J.C., Burton, D., Wilson, I.A., Wong, C.-H. Covalent display of oligosaccharide arrays in microtiter plates. J. Am. Chem. Soc. 126:8640, 2004. Collins, B.E., Paulson, J.C. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr. Opin. Chem. Biol. 8:617, 2004. Goldberg, D., Sutton-Smith, M., Paulson, J.C., Dell, A. Automatic annotation of matrix-assisted laser desorption/ionization N-glycan spectra. Proteomics 5:865, 2005. Han, S., Collins, B.E., Bengtson, P., Paulson, J.C. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat. Chem. Biol. 1:93, 2005. Ikehara, Y., Ikehara, S.K., Paulson, J.C. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J. Biol. Chem. 279:43117, 2004. Kitazume, S., Nakagawa, K., Oka, R., Tachida, Y., Ogawa, K., Luo, Y., Citron, M., Shitara, H., Taya, C., Yonekawa, H., Paulson, J.C., Miyoshi, E., Taniguchi, N., Hashimoto, Y. In vivo cleavage of α2,6-sialyltransferase by Alzheimer’s β-secretase. J. Biol. Chem. 280:8589, 2005. Vyas, A.A., Blixt, O., Paulson, J.C., Schnaar, R.L. Potent glycan inhibitors of myelin-associated glycoprotein enhance axon outgrowth in vitro. J. Biol. Chem. 280:16305, 2005. Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved. 237 Published by TSRI Press®. © Copyright 2005, The Scripps Research Institute. All rights reserved.